

 Ajouter à une liste de lecture
Ajouter à une liste de lecture
Dans les précédents articles [1, 2], nous avions découvert quelques outils d’analyse bioinformatique et procédé à une première analyse d’une région d’intérêt du virus SARS-CoV-2. Nous allons avec ce dernier opus finir d’explorer la structure de cet organisme et illustrer encore une fois l’incidence du modèle linuxien sur les progrès dans les sciences.
Résumé des épisodes précédents [1,2] : à partir de la séquence de référence du virus SARS-CoV-2 (NC_045512.2), nous avons détecté 191 régions potentiellement à l’origine des protéines que le virus fait synthétiser par son hôte pour pouvoir se reproduire. De ces 191 régions, sur la base de critères assez basiques, nous en avons sélectionné 36 a priori intéressantes. Nous avons mené quelques investigations sur la première de ces régions et montré qu’elle correspondait à une protéine connue chez d’autres coronavirus, à la base de leur capacité à se reproduire.
Nous allons aujourd’hui généraliser la mécanique de notre étude pour l’appliquer aux 35 régions restantes. Et comme annoncé dans le précédent article, si vous prenez le train en route, tout n’est pas perdu (cf. note) !
Les données récupérées, les résultats obtenus et les scripts développés dans le cadre de cette série d’articles sont disponibles sur la forge logicielle GitLab hébergée par Framasoft : https://framagit.org/doccy/SARS-CoV-2-bioinfo-analysis.
1. Rappel des étapes d’analyse
La première région que nous avions étudiée correspondait au 81e ORF (Open Reading Frame – cadre ouvert de lecture) dont la traduction sous forme de séquence d’acides aminés est dans le fichier results/ORF/nc_045512.2_81.fasta.
1.1 La démarche qui a marché
Cette séquence a été confrontée à la base de données UniProtKB/Swiss-Prot(swissprot) via l’outil BLAST [3] (nous utilisons la version pour les protéines) disponible en ligne sur le site du NCBI [4]. Pour cela, il a suffi de téléverser la séquence dans le formulaire, de sélectionner la base de données, d’exclure les informations relatives au SARS-CoV-2 pour le fairplay (puisque nous étudions l’organisme comme s’il venait juste d’être découvert), puis de cliquer sur le bouton de soumission.
Après quelques instants, nous avons récupéré les 100 séquences (limite par défaut) les plus significativement proches et nous avons sélectionné les séquences qui s’alignaient sur plus de 80 % de notre séquence (en triant par ordre décroissant au niveau de la colonne query cover). Nous les avons téléchargées (menu déroulant download) au format GenBank dans le fichier results/ORF/nc_045512.2_81_related.gb. À partir de cet ensemble, nous avons construit un fichier contenant toutes ces séquences ainsi que notre séquence requête, puis nous avons réalisé un alignement multiple avec le programme ClustalΩ (en passant par son wrapper emma fourni par la suite EMBOSS [5,6]). Nous avons généré quelques images, notamment via l’outil plotcon de la suite EMBOSS (qui illustre le degré de conservation sur une fenêtre glissante d’un alignement) et les outils newicktotxt et newicktops du programme NJplot (qui permettent de montrer l’arbre de guidage généré lors de l’alignement ; si la séquence requête est éloignée des autres séquences, cela se voit). Ceci nous a permis de constater que les séquences se divisaient en deux groupes, d’un côté les séquences concernant une protéine appelée Replicase polyprotein 1ab, et de l’autre celles concernant une protéine appelée Replicase polyprotein 1a. Notre séquence requête se situant visiblement dans ce second groupe, nous avons réitéré la même analyse en ne considérant que les séquences codant pour la Replicase polyprotein 1a. Cette fois-ci, l’alignement produit a été bien plus concluant et nous a permis d’établir avec confiance que notre ORF pouvait être considéré de facto comme une CDS (Coding Data Sequence) associée au gène de la Replicase polyprotein 1a. Ceci est d’autant plus convaincant que les séquences récupérées par BLAST correspondent toutes à des coronavirus et que la Replicase polyprotein 1a est une sorte de super protéine qui permet aux virus qui en disposent de se répliquer.
L’automatisation de ces étapes consiste en deux parties, d’une part faire la recherche de séquences proches dans la base de données UniProtKB/Swiss-Prot(swissprot) avec BLAST en ligne, et d’autre part dérouler les programmes à la suite les uns des autres.
1.2 La recherche des séquences proches
La bonne nouvelle, c’est que BLAST nous autorise à soumettre plusieurs séquences en une fois. Donc finalement, la partie téléversement et paramétrage n’a pas besoin d’être automatisée.
Il faut donc créer un fichier contenant nos séquences, ce qui ne pose pas de problème majeur vu que nous disposons de toutes nos séquences individuelles dans le répertoire results/ORF/ et qu’elles ont toutes le même préfixe nc_045512.2_ suivi du numéro de l’ORF suivi de l’extension .fasta. Notez au passage l’utilisation de l’option --sort=version qui permet de trier les fichiers comme s’il s’agissait de numéros de version. Ainsi le fichier nc_045512.2_81.fasta est bien avant le fichier nc_045512.2_106.fasta :
Pour la soumission, il suffit donc de téléverser ce fichier, de sélectionner la base de données UniProtKB/Swiss-Prot(swissprot) et d’exclure les informations relatives au SARS-CoV-2 (cf. figure 1).
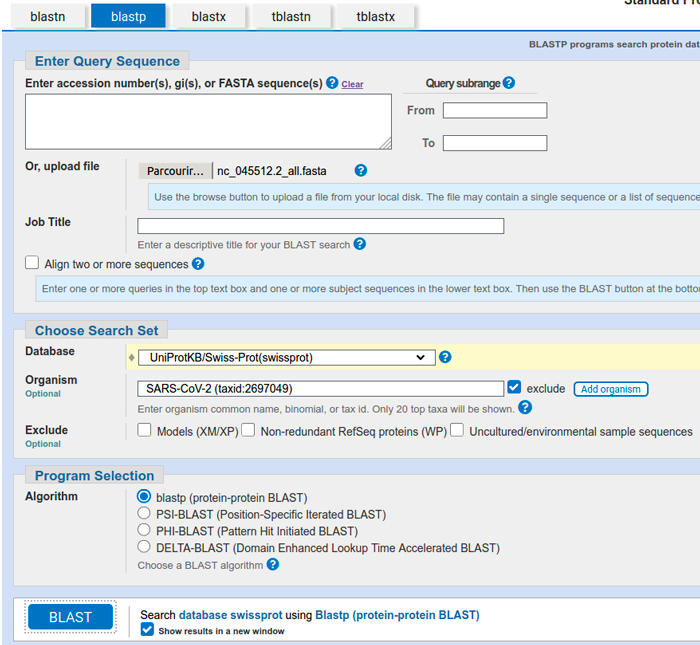
Une fois la demande soumise, nous obtenons assez rapidement tous les résultats de nos requêtes. Pour sélectionner les résultats d’une requête, il suffit d’aller dans le menu déroulant associé à la ligne Results for.
Les résultats précédés d'un astérisque sont ceux pour lesquels aucune séquence semblable n'a été retrouvée. Sur les 36 ORF, il y en a 22 qui ne donnent aucun résultat (3, 16, 24, 36, 37, 43, 58, 80, 82, 83, 94, 106, 122, 139, 160, 162, 171, 174, 188, 189, 190 et 191). Pour les 14 autres (81, 137, 159, 167, 168, 169, 172, 173, 175, 176, 179, 180, 183 et 187), il y a des résultats. Le premier est l’ORF n°81 que nous avons déjà étudié et pour lequel nous retrouvons les mêmes séquences (fort heureusement).
Commençons par étudier les ORF pour lesquels nous n'avons pas trouvé de résultats et calculons les tailles des ORF et des chaînes d'acides aminés qui en résulteraient. Pour cela, nous partons du fichier GFF que nous avions produit la dernière fois (results/nc_045512.2_ORF_filtered.gff) et qui n’est autre qu’un fichier tabulé contenant soit des commentaires ou des métadonnées commençant par un # (c’est un dièse, pas un hashtag !), soit des lignes dont le 6e champ correspond à la taille de l’ORF + 3 (pour le codon stop [1]) et le 9e champ a systématiquement – parce que nous l’avons écrit ainsi – la forme ID=nc_045512.2_<ID>; name=ORF_<ID> (où <ID> correspond au numéro de l’ORF). Ainsi, il suffit d’écrire une commande awk qui consiste à afficher, au démarrage, nos entêtes de colonnes ; puis pour chaque ligne ne commençant pas par un # dans le fichier GFF, l’identifiant obtenu en supprimant tout ce qui précède le dernier _ du 9e champ, puis la valeur diminuée de 3 du 6e champ et enfin cette valeur divisée par 3 puisqu’il faut 3 nucléotides pour coder un acide aminé. En passant, le résultat de cette commande à la commande sort (avec l’option -n), nous obtenons la liste des ORF, de leur longueur et de la longueur des chaînes qu’ils pourraient produire, dans l’ordre de leurs identifiants :
Nous allons simplement commenter (avec un #) les ORF pour lesquelles BLAST a donné un résultat et nous pouvons passer rapidement ce fichier pour analyse à gnuplot qui sait très bien faire des statistiques :
Cette rapide statistique permet de voir que les tailles des ORF non retrouvés vont de 30 nucléotides (10aa – acides aminés) à 261 nucléotides (87aa), la moitié faisant moins de 153 nucléotides (51aa) et les trois quarts faisant moins de 177 nucléotides (59aa), donc il n'y a rien de surprenant à ce que ces ORF n'aient pas été retrouvés dans la base de données UniProtKB/Swiss-Prot(swissprot) et il est peu probable que ceux-ci soient réellement codants pour des [parties de] protéines.
Nous pouvons donc sereinement attaquer l’analyse des ORF pour lesquels nous avons des résultats.
1.3 Le script du bourrin
Vu que nous allons essentiellement exécuter les outils que nous avions testés en ligne de commande, le Bash est tout indiqué ici. Nous allons partir du principe que le script, que j’ai baptisé brutal_analysis.sh, n’a besoin que de l’identifiant de l’ORF pour dérouler la suite des outils. Cependant, nous n’allons pas prendre trop de risques non plus et commencer par dire au script de cesser immédiatement dès qu’une erreur se produit (ligne 3). Nous allons récupérer le répertoire de travail en nous basant sur le chemin menant au script (ligne 5) et vérifier que la variable n’est pas vide par acquit de conscience (ligne 6). Nous vérifions également que nous pouvons nous rendre dans le répertoire hébergeant le script et que nous pouvons lire son contenu (lignes 8 à 15).
Nous récupérons l’identifiant de l’ORF à étudier qui doit être le premier argument de notre script et nous nous assurons que cet identifiant a bel et bien été fourni en ligne de commande, sans quoi nous affichons un message rudimentaire sur l’utilisation de ce programme.
Nous remontons d’un niveau dans l’arborescence des répertoires (donc a priori dans le répertoire analysis), puis définissons quelques variables pour rendre le script potentiellement évolutif et éviter les coquilles typographiques qui font pleurer :
Ainsi, fich_tpl devrait valoir quelque chose comme results/ORF/nc_045512.2_${ORF_ID}_related et tmp_dir (répertoire temporaire) devrait valoir quelque chose comme results/tmp_${ORF_ID}.
Nous créons donc le répertoire temporaire (lignes 35 et 46).
Lorsque l’on télécharge les séquences sélectionnées dans BLAST, mon navigateur me propose de l’enregistrer sous le nom sequences.gb. Cela me convient très bien, j’enregistrerai les fichiers dans le répertoire /tmp. Il me suffit donc de déplacer ce fichier dans le répertoire de travail sous son nom définitif (ligne 39), puis d’extraire les séquences dans le répertoire temporaire (ligne 42), ainsi que les annotations qui vont avec (ligne 44). Cela nous permet de créer le fichier contenant toutes les séquences, y compris la séquence de l’ORF en cours d’analyse (ligne 47).
Nous procédons à l’alignement des séquences (ligne 50), à la génération des représentations des arbres de guidage (lignes 53 à 57) et lançons son affichage avec EOG ou à défaut le programme associé aux fichiers PNG (ligne 58).
Nous créons le graphique de conservation de l’alignement (ligne 61) que nous visualisons directement (ligne 64) afin de pouvoir vérifier aussi que l’alignement est représentatif (que les séquences sont suffisamment semblables pour pouvoir transférer les informations).
Comme rien ne vaut malgré tout un regard sur l’alignement brut, nous l’ouvrons avec le pager less.
Enfin, selon la confiance que l’alignement nous inspire, il reste à copier-coller les annotations qui nous semblent pertinentes (car retrouvées dans toutes les séquences) en vue de transformer notre ORF en CDS et en gène. Pour ma part, j’utilise joe qui présente tous les inconvénients de vim et emacs réunis sans avoir un seul de leurs avantages (bref, j’adore !).
Nous pouvons vérifier rapidement que la requête correspondant à l'ORF 81 (que nous avions traitée) donne les mêmes résultats (en sélectionnant les « bonnes » séquences). Parfait… analysons en masse.
1.4 Analyse du berserker
La mécanique est simple, pour chacune des requêtes, nous récupérons les séquences qui couvrent au moins 80 % de la requête dans le fichier /tmp/sequence.gb et nous appliquons le script précédent en lui passant le numéro de l'ORF en argument.
Par exemple, pour la seconde requête ayant donné un résultat (15e séquence requête, qui correspond à l’ORF n°137 ; cf. figure 2), nous retrouvons en premier les séquences provenant des mêmes organismes que précédemment (des coronavirus infectieux de la chauve-souris) et codant toutes pour la protéine Replicase polyprotein 1ab.
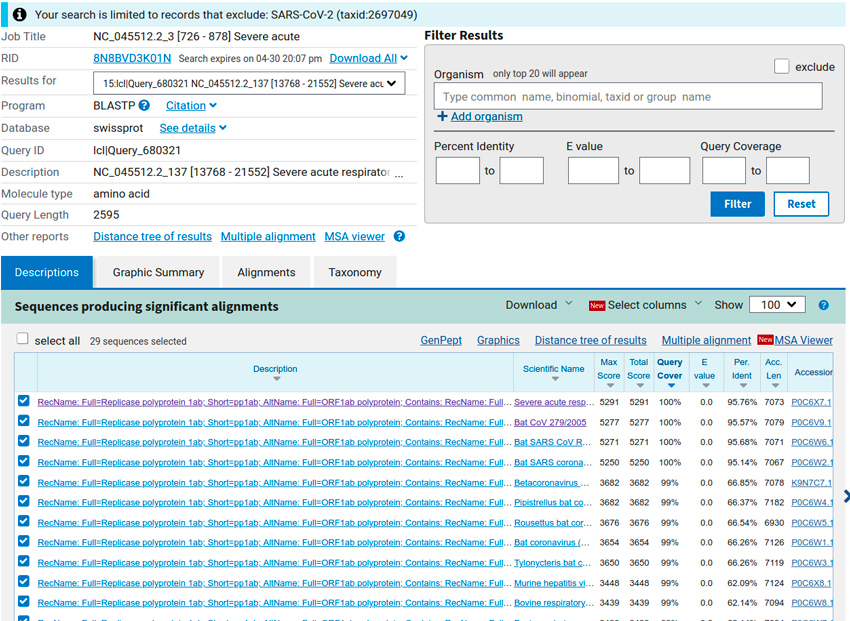
L'alignement montre clairement que la requête s'aligne seulement à la fin des autres séquences. Or pour l’ORF précédent (n°81), nous avions vu que sur ces mêmes séquences, l’alignement était partiel et portait seulement sur le début des séquences. Il ne semble pas exister de protéine Replicase polyprotein 1b, mais [7] explique que lorsque la séquence codant la Replicase polyprotein 1a est suivie d'un ORF valide, éventuellement avec un décalage de cadre de lecture, ce qui est le cas ici, le produit de cet ORF peut se combiner visiblement avec la chaîne d’acides aminés de la protéine Replicase polyprotein 1a pour former la protéine Replicase polyprotein 1ab. Ceci corrobore le résultat obtenu pour l'ORF n°81 que nous avions détecté comme étant codant pour la Replicase protein 1a.
En appliquant le script, nous réussissons à annoter tous les ORF restants sans aucune difficulté, excepté pour l'ORF n°168, où il n'y a qu'une seule séquence résultat. Ceci empêche le bon déroulement de l'étape d'alignement multiple (qui requiert de devoir aligner au moins 3 séquences), mais une astuce assez simple consiste à dupliquer le contenu du fichier /tmp/sequence.gb avant de lancer le script. Ceci étant, l'alignement est vraiment très partiel et il est difficile de déduire une information probante.
Il semble évident au vu des séquences retrouvées pour chacun des ORF que la séquence étudiée est un coronavirus proche de ceux qui infectent les chauves-souris. Il reste quelques ORF pour lesquels nous n’avons pas eu d’informations.
Pour les très petits ORF (ceux qui codent pour moins de 30 acides aminés), nous pouvons nous contenter de les supprimer sans scrupule, car il y a peu de chances qu'ils codent réellement pour des protéines. Cela concerne les ORF numéros : 82 (13aa), 83 (24aa), 174 (12aa), 188 (10aa), 190 (11aa) et 191 (12aa). Nous pouvons laisser les annotations des autres ORF dans le doute.
Il est également possible (voire même judicieux) d’ajuster le début de la région 3'-UTR à partir de la dernière région codante que nous avons détectée.
Ceci nous donne le fichier final suivant (results/nc_045512.2.gff) :
Il est bien évidemment possible de pousser l'analyse davantage, mais nous nous arrêterons là pour cette séquence.
2. Qu’a-t-on découvert ?
Produire un beau fichier GFF, c’est sympa, mais ça laisse tout de même un peu perplexe. Nous pouvons dans un premier temps essayer de le visualiser.
2.1 Le so-ASCII (so cute)
La suite EMBOSS propose le programme showfeat pour effectuer un rendu ASCII des annotations sur une séquence, mais ce programme n’accepte pas le GFF. Qu’à cela ne tienne, créons une séquence annotée au format EMBL :
Nous découvrons un autre format, mais le contenu est relativement bien préservé, si ce n’est que les lignes commençant par FT sont mal formées, car elles contiennent l'identifiant de séquence avant les positions et ça, showfeat n’aime pas.
Ceci n’est pas trop grave, un coup de sed et c’est fixé :
Nous pouvons à présent afficher la séquence annotée (c’est trop stylé) :
Il y a d’autres visualisations possibles par exemple avec showseq qui propose différents formats (ici, j’ai sélectionné l’affichage avec traduction en séquence d'acides aminés) :
2.2 Et à part ça ?
Il est toujours intéressant de regarder une véritable image. Ainsi la figure 3 a été produite au moyen d’annotation sketch de la suite GenomeTools [8].
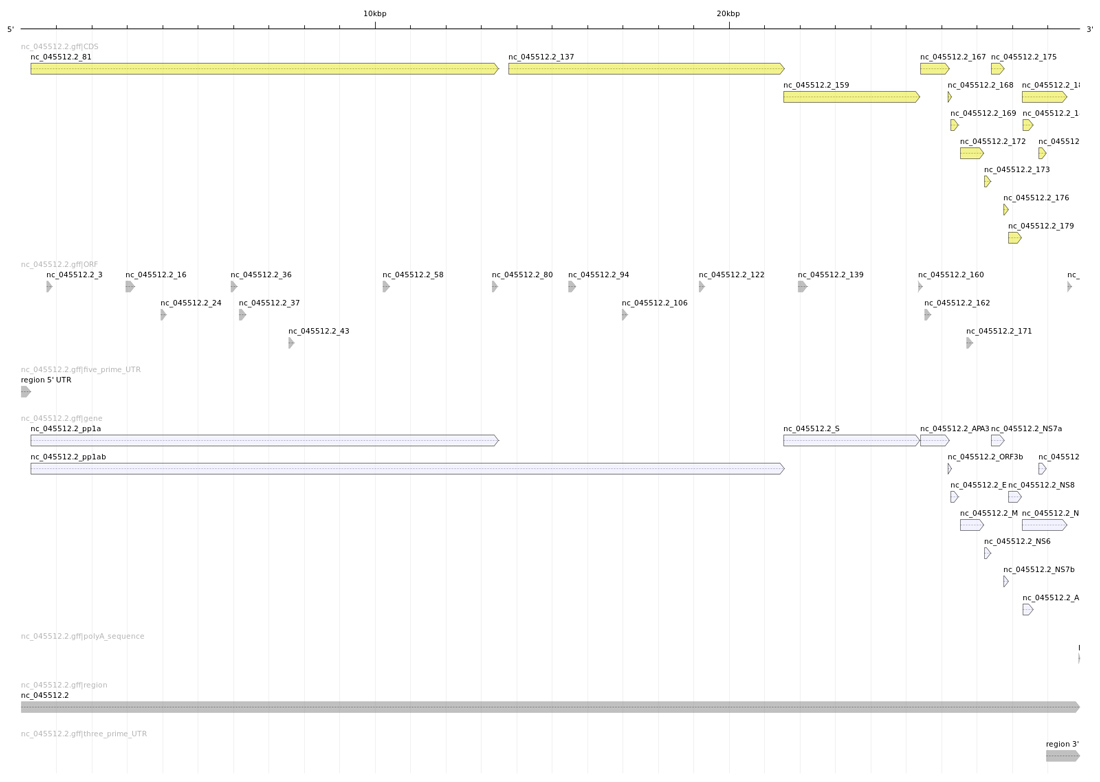
Ce qui est encore plus intéressant, c’est de comparer cette image avec celle produite avec le fichier GFF officiel des annotations de SARS-CoV-2 (disponible sur le site du NCBI et dans le dépôt Git, cf. figure 4).
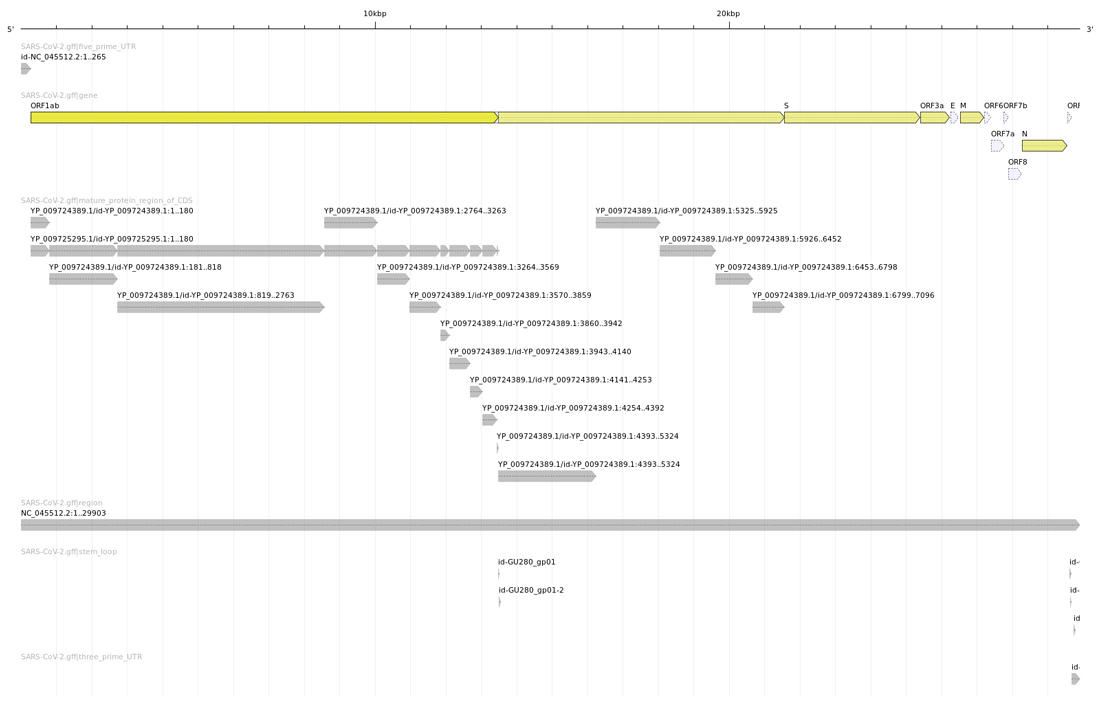
Nous nous apercevons que notre analyse assez simpliste est très proche de la réalité (au sens biologique du terme). Nous retrouvons en premier lieu les deux Replicase polyprotein 1a (ORF 81) et Replicase polyprotein 1ab (ORF 81 & 137) permettant au SARS-CoV-2 de se reproduire ; la Spike glycoprotein (ORF 159) permettant au virus de s’accrocher aux membranes des cellules hôtes et de les pénétrer ; l’Enveloppe small membrane protein (ORF 169) et la Membrane protein (ORF172) permettant au virus de produire la membrane de sa capside (sa coque), la Nucleoprotein (ORF 187) qui participe à sa réplication et à sa transcription par l’hôte. Tous ces éléments sont essentiels au cycle de vie du virus (oups ! Il paraît que les virus ne sont pas vivants) et font bien évidemment l’objet de nombreuses études en vue de pouvoir lutter efficacement contre sa propagation, puisqu’ils permettent de comprendre la structure de cet organisme (cf. figure 5).
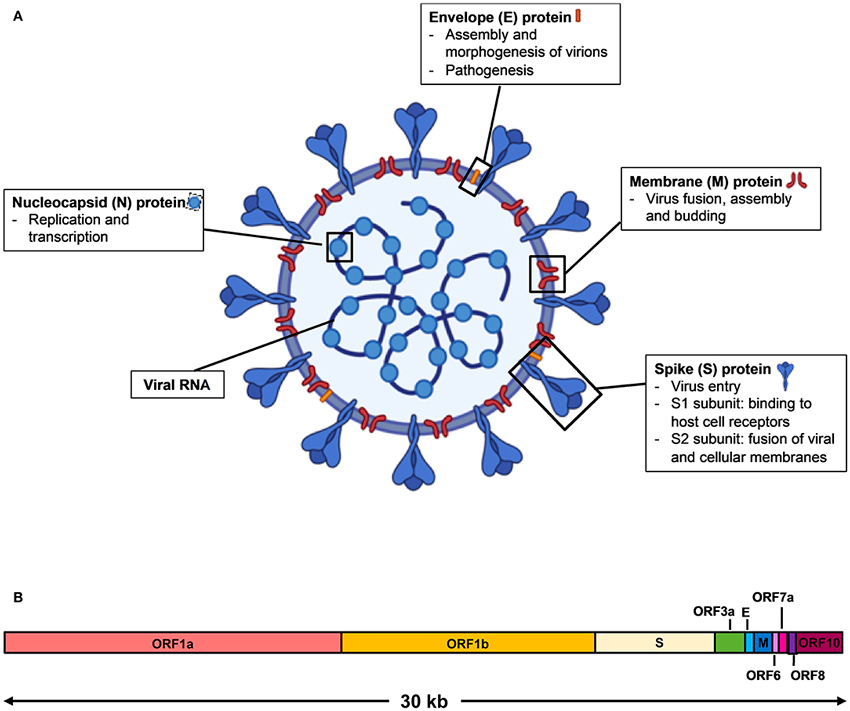
Imaginons par exemple que seule la séquence codante de la protéine Spike glycoprotein (qui est présente en surface de la capside) soit injectée dans des cellules d’un hôte potentiel. Les cellules de l’hôte vont synthétiser cette protéine et le système immunitaire pourra donc s’entraîner à la reconnaître et ainsi apprendre à lutter contre l’envahisseur fictif. Ainsi, en cas de contamination par le virus, l’hôte saura se défendre aussitôt et endiguer la prolifération du virus. C’est le principe des vaccins à ARN. Cette stratégie est robuste tant qu’il n’y a pas de mutations importantes affectant la séquence codant cette protéine (sinon il faut vacciner à nouveau avec la séquence mutée).
Conclusion
Vous l’aurez compris, cette analyse est très partielle et manque de rigueur. Toutefois, elle permet de montrer une fois de plus l’apport du logiciel libre (open source) et l’intérêt de l’ouverture des données (open data). Bien entendu, dans un véritable cadre d’analyse, les scripts mis en place ne sont pas suffisants (du point de vue méthodologique), pas suffisamment documentés, pas assez robustes (absence de tests), etc.
La plupart des unités de service en bioinformatique (en réalité toutes celles que je connais) travaillent exclusivement sous GNU/Linux. Ce n’est pas par idéologie, mais uniquement parce que c’est le seul environnement qui permet de travailler avec de bons outils en toute sérénité.
Étonnamment, à l’heure où tout le monde découvre le Big Data et où certains groupes tentent de s’imposer dans le secteur, en bioinformatique (où le volume de données croît exponentiellement depuis plus de 15 ans), la quasi-totalité des outils d’analyse développés le sont dans un cadre académique, avec parfois peu de moyens et de support. Ceci explique qu’une grande partie des outils sont finalement des prototypes aboutis plutôt que de réels logiciels. Et pourtant, ils permettent d’appréhender (efficacement) et de mieux comprendre le domaine du vivant.
Références
[1] MANCHERON A., « Voyage initiatique vers la bioinformatique : les premiers pas », GNU/Linux Magazine France n°251 : https://connect.ed-diamond.com/gnu-linux-magazine/glmf-251/voyage-initiatique-vers-la-bioinformatique-les-premiers-pas
[2] MANCHERON A., « Voyage initiatique vers la bioinformatique : en route pour l’aventure », GNU/Linux Magazine France n°253 : https://connect.ed-diamond.com/gnu-linux-magazine/glmf-253/voyage-initiatique-vers-la-bioinformatique-en-route-pour-l-aventure
[3] S. F. ALTSCHUL, W. GISH, W. MILLER, E. W. MYERS et D. J. LIPMAN, « Basic Local Alignment Search Tool », Journal of molecular biology, n°215(3),1990, p 403 à 410.
[4] Site du National Center for Biotechnology Information (centre national américain) : https://www.ncbi.nlm.nih.gov/
[5] P. RICE, I. LONGDEN et A. BLEASBY, « EMBOSS: The European Molecular Biology Open Software Suite », Trends in Genetics n°16(6), 2000, p 276 à 277.
[6] Site officiel de la suite EMBOSS : http://emboss.sourceforge.net/
[7] J. ZIEBUHR, « The Coronavirus Replicase - Coronavirus Replication and Reverse Genetics », Current Topics in Microbiology and Immunology, n°287, 2005.
[8] Site officiel de la suite GenomeTools : http://genometools.org/
[9] C. Y.-P. LEE, R. T. P. LIN, L. RENIA et L. F. P. NG, « Serological Approaches for COVID-19 : Epidemiologic Perspective on Surveillance and Control », Frontiers in Immunology, 2020.
Pour aller plus loin
Nous avons traité dans cette série d’articles d’analyse de séquences et de génomique comparative (deux des domaines de la bioinformatique, parmi tant d’autres). Nous nous sommes restreints à l’analyse des structures dites primaires (dans une seule dimension). Il serait tout à fait possible de poursuivre cette étude en essayant de modéliser les structures secondaires de nos séquences (les conformations locales en 3D de leurs différentes parties), puis de leurs structures tertiaires (agencement dans l’espace des structures secondaires) et quaternaires (interactions des structures entre elles). Si cette petite balade dans l’univers de la bioinformatique vous a intéressés, sachez qu’il y a de la place pour toutes et tous, que vous soyez pythoniste convaincu, développeur C chevronné, bayésien né, théoricien des graphes ou encore adeptes de l’apprentissage automatique (ce qui évite de parler d’intelligence, même si on la qualifie d’artificielle :D). D’accord, il faut s’intéresser d’un peu plus près à la biologie, mais sachez que les spécialistes que j’ai rencontrés jusqu’à présent m’ont toujours transmis avec patience, bienveillance et simplicité les connaissances nécessaires à l’exercice de mon métier ; tous sans aucune exception ! Alors je vous invite très cordialement à aller plus loin.



