

 Ajouter à une liste de lecture
Ajouter à une liste de lecture
Dans un précédent article [1], nous avions découvert la suite EMBOSS et mis en évidence des régions d’intérêt de la séquence nucléique de référence du virus SARS-CoV-2. Nous allons donc continuer le chemin en exploitant de nouveaux outils, certains issus de la bioinformatique et d’autres issus de la communauté linuxienne.
Où en étions-nous ? Ah oui, je me souviens…
Nous avions récupéré quelques séquences de SARS-CoV-2, dont la séquence de référence du virus (NC_045512.2). Nous avions également installé et pris en main la suite EMBOSS [2,3], ce qui nous avait permis de commencer l’analyse des séquences récupérées. Nous avions notamment mis en évidence des régions de la séquence de référence, appelées ORF (Open Reading Frames – cadres ouverts de lecture), qui correspondent à de potentielles protéines que le virus ferait produire par l’organisme hôte (celui qu’il squatte sans vergogne) et qui lui permettent de se reproduire. Ah oui, j’avais oublié. La biologie est ici présentée sous un angle assez imagé et peut parfois piquer les yeux des puristes (donc, si vous avez dans votre entourage des biologistes, n’essayez pas de frimer en reprenant mes propos, cela pourrait tourner à votre désavantage). Par exemple, vous pouvez tout à fait vous représenter un virus comme une bombe fork. Son seul objectif est de se reproduire et ça finit par faire planter le système, si celui-ci ne parvient pas à le détecter et à l’éradiquer (en l’occurrence, dans le cas du SARS-CoV-2, c’est plutôt le système qui « exabuse » – du verbe exabuser – la contre-attaque).
Vous aviez tout bien fait la dernière fois, mais vous ne savez plus où sont les résultats que vous aviez obtenus ! Ne vous inquiétez pas, nul besoin de tout refaire (cf. note).
Les données récupérées, les résultats obtenus et les scripts développés dans le cadre de cette série d’articles sont disponibles sur la forge logicielle GitLab hébergée par Framasoft : https://framagit.org/doccy/SARS-CoV-2-bioinfo-analysis.
1. Régions d’intérêt de la séquence de référence
Nous avions récupéré les ORF valides dans le fichier results/nc_045512.2_ORF.fasta partiellement reproduit ci-après :
Pour chaque ORF, nous disposons d’un nom de séquence qui est le nom d’origine de la séquence suffixé d’un identifiant (compteur commençant à 1) ; de ses positions dans la séquence (information entre crochets) ; du descriptif associé à la séquence d’origine et de la séquence d’acides aminés correspondant à sa traduction. Nous avions également mentionné que le dernier ORF contenant un codon start (une Méthionine), le programme getorf avait renvoyé la traduction de la fin de la séquence à partir de la position 29 866 ; cependant, cet ORF n'est pas valide en réalité, c'est juste qu'il va jusqu'à la fin de la séquence.
Avant toute chose, commençons par récupérer les coordonnées des régions codantes potentielles. Pour cela, il suffit de récupérer les lignes du fichier qui commencent par > (avec grep), puis à capturer l’identifiant, la position de début et la position de fin de l’ORF (avec sed), tout conserver sauf la dernière (avec head) et enfin rediriger dans le fichier, tout en l’affichant sur la sortie standard (avec tee) :
À partir de maintenant, les choses sérieuses peuvent commencer.
1.1 Visualisation des ORF
La bioinformatique admet de très nombreuses définitions (et je n’entrerai pas dans le débat, même si j’ai un avis assez tranché sur la question), cependant il faut comprendre que l’usager des outils développés reste pour une grande majorité un biologiste. Donc, son expertise passera à un moment ou à un autre par une représentation graphique.
Notre objectif est d’annoter des portions de séquences, mais également de permettre de visualiser le produit de nos annotations. Il existe plusieurs formats pour représenter les annotations, parmi eux le format GFF (voir encadré) qui, outre son format assez facile à exploiter sous GNU/Linux, établit un lien direct entre l’annotation et la Sequence Ontology [4] donnant ainsi à la fois un formalisme rigoureux et une véritable sémantique aux annotations.
Le format GFF
Le « Generic Feature Format » permet d’annoter tout ou parties des séquences biologiques. Il s’agit simplement d’un fichier tabulé où chaque ligne représente une annotation. Il y a neuf champs (séparés donc par des tabulations) pour chaque annotation.
|
Champ |
Nom |
Description |
|
1 |
séquence |
Nom (identifiant) de la séquence porteuse de l’annotation. |
|
2 |
source |
Identifiant de la source de l’annotation (nom du programme, de la base de données, du consortium…). |
|
3 |
type |
Type de l’élément annoté qui doit correspondre à un des termes de la Sequence Ontology [4]. |
|
4 |
début |
Position de début de l’annotation sur la séquence (la numérotation commence à 1). |
|
5 |
fin |
Position (incluse) de fin de l’annotation sur la séquence (la numérotation commence à 1). |
|
6 |
score |
Fiabilité de l’annotation (lorsque celle-ci est prédite par un outil, elle est souvent assortie d’une métrique représentative de la fiabilité de la prédiction). L’absence de score est dénotée par un .. |
|
7 |
brin |
Indication du brin codant de l'élément (voir [1]). Ce champ admet trois valeurs possibles : + pour le sens de lecture « normal », - pour le sens de lecture inverse et . lorsque l’information est indéterminée. |
|
8 |
phase |
Phase du cadre de lecture applicable aux annotations de type CDS (voir [1]). Celle-ci peut prendre 4 valeurs : 0, 1 ou 2 si le type de l’annotation est CDS et . dans les autres cas. |
|
9 |
attributs |
Autres informations relatives à l’annotation. |
Le dernier champ est donc celui qui permet d’écrire un peu ce que l’on veut. Il est généralement composé d’une suite de couples clé=valeur séparés par des ;. Il est recommandé d’utiliser le mécanisme d’encodage de la RFC 3986 relative à la syntaxe des URI (Uniform Resource Identifier) [5] pour les caractères devant être échappés (tabulation, nouvelle ligne, retour chariot, #, %, ;, =, &, ,, etc.). C’est-à-dire d’utiliser le symbole % suivi du codage hexadécimal à deux symboles correspondant au code ASCII du caractère échappé (p. ex., le % est représenté par %25 et le ; par %3B). Il est suggéré d’utiliser l’encodage UTF‑8 pour le reste (cette préconisation est une hérésie pour l’informaticien que je suis, mais pleine de bon sens pour le bioinformaticien que je suis également).
Le format GFF autorise également l’utilisation du # en début de ligne pour marquer le début d’un commentaire allant jusqu’à la fin de la ligne.
Enfin, il est possible de fournir des métadonnées. Ces métadonnées sont des lignes qui commencent par ##. La plus importante est celle permettant d’indiquer la version de la spécification de GFF utilisée dans le fichier (p. ex., ##gff-version 3.1). Cette information doit figurer en première ligne du fichier. Les autres métadonnées disponibles dépendent de la version utilisée. On retrouve parfois des métadonnées officieuses utilisées par certains outils. Ces métadonnées commencent par #! et sont donc ainsi assimilables à des commentaires.
Nous allons donc écrire un script awk permettant de transformer notre fichier CSV en GFF. Pour cela, nous allons représenter les ORF sous forme de tableaux à trois clés (id, deb et fin). Outre une fonction permettant de renvoyer la taille d’un ORF (lignes 17 à 19), comme je ne sais pas définir une fonction qui renvoie un tableau en awk (je ne suis pas certain que cela soit possible, par ailleurs), je contourne la difficulté en passant par un procédé de sérialisation/dé-sérialisation (lignes 24 à 26 et 33 à 37) :
Au début du programme, il ne se passe pas grand-chose (lignes 41 à 43), mais à chaque début de fichier passé en ligne de commande (lignes 47 à 57), nous remettons le compteur d’ORF à 0 (variable nb), nous initialisons la variable name au nom du fichier sans l’extension (ce sera notamment le champ 1 du GFF produit) et nous réinitialisons la position maximale de la séquence pour le fichier en cours à 1 (variable max_pos).
Pour chaque ligne du fichier en cours de lecture (lignes 60 à 68), il suffit d’associer à l’ORF en cours les valeurs de chaque champ à chaque clé (notons que nous ajoutons 3 à la borne de fin, car les positions renvoyées par getorf n’incluent pas le codon stop). Nous en profitons pour mettre à jour la valeur de la plus grande position.
Arrivés à la fin de la lecture du fichier en cours (lignes 71 à 78), nous affichons les métadonnées associées au fichier GFF (la position maximale est une estimation), puis pour chaque ORF, nous produisons une annotation de type ORF (champ 3). Nous utilisons la taille de l’ORF en guise de score (champ 6). Nous en profitons pour calculer la phase de lecture (champ 8 de l’annotation).
En appliquant ce script (après l’avoir rendu exécutable) au fichier CSV, nous obtenons :
Comme la position de fin de la séquence est une estimation, nous allons corriger cela rapidement :
Il existe un outil permettant de visualiser les fichiers GFF. Il s’agit du programme annotation sketch de la suite GenomeTools [6] qu’il est possible d’installer localement (par exemple, sous Debian/Ubuntu) :
On peut l’utiliser en ligne de commande (commande gt sketch), mais il est également possible d’utiliser l'outil en ligne à l’adresse http://genometools.org/cgi-bin/annotationsketch_demo.cgi (ce que j’ai fait pour cet article en renseignant, pour plus de confort visuel, une largeur de 1600 pixels au lieu des 800 pixels proposés par défaut ; cf. figure 1).

Visuellement, nous nous apercevons aisément que tous les ORF ne semblent pas présenter le même intérêt. Sans entrer dans les détails de la biologie, une protéine est généralement composée en moyenne de 300 à 400 acides aminés, et très rarement moins de 100 acides aminés. Nous allons donc tenter de déterminer des critères permettant de filtrer les ORF à étudier.
1.2 Sélection des ORF candidats
Même s’il paraît que ce n’est pas la taille qui compte, intéressons-nous de plus près à la distribution des longueurs des ORF.
Pour cela, nous pouvons écrire un petit script gnuplot assez simpliste :
Il reste à le rendre exécutable et à le lancer pour obtenir l’histogramme présenté en figure 2.
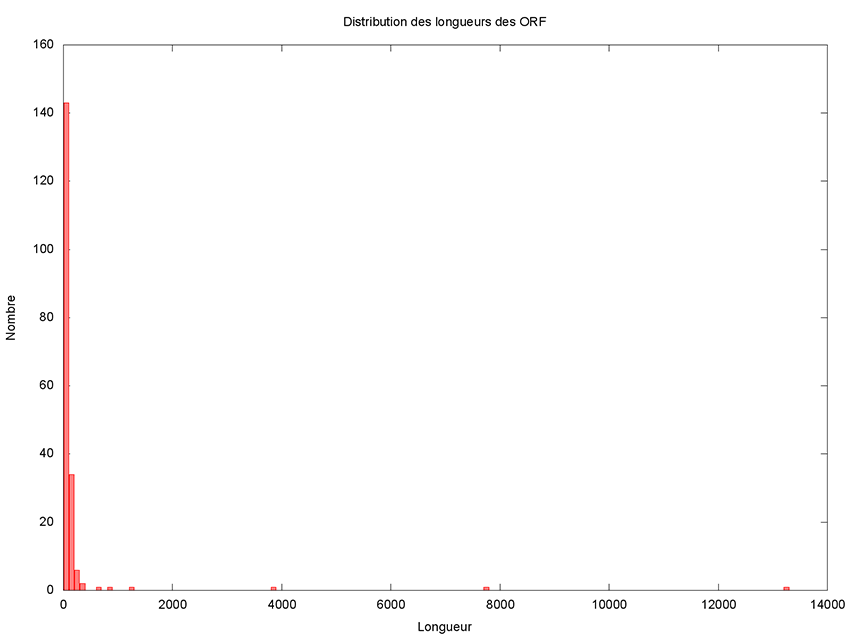
Cet histogramme fait clairement apparaître 5 grands (voire très grands) ORF candidats. Il est donc très raisonnable de ne pas considérer les petits ORF qui seraient inclus dans des plus grands. Un ORF de 150 nucléotides code pour 49 acides aminés plus 1 codon stop, ce qui est vraiment très petit et donc est un critère de filtrage très satisfaisant. Copions le script précédent dans un nouveau script appelé filtre_ORF.awk et ajoutons donc une variable permettant de définir le seuil en dessous duquel nous considérerons un ORF comme petit.
Cette fonction d’initialisation devra être appelée au démarrage du script.
Il nous faut également une fonction permettant de tester si un ORF en inclut un autre.
Pour pouvoir faire le filtrage, la stratégie gloutonne mise en place est assez simple (et non optimale). Elle consiste pour chaque ORF, du plus grand au plus petit, à supprimer les ORF inclus et considérés comme petits. Il nous faut donc pouvoir trier les ORF par tailles décroissantes. Sachant que awk propose une fonction asort(src, dst, cmp) permettant de remplir le tableau dst à partir des données du tableau src dans l’ordre défini par la fonction cmp. La fonction cmp doit prendre 4 paramètres (la position du premier élément à comparer dans le tableau src, ce premier élément, la position du second élément à comparer dans le tableau src et enfin ce second élément). Cette fonction doit renvoyer -1 si le premier élément précède le second élément, 1 dans le cas contraire et 0 si ces deux éléments sont équivalents. Ceci aboutit à la définition de la fonction de comparaison de deux ORF en fonction de leur taille :
Une fois que les ORF auront été filtrés, nous les réordonnerons dans l’ordre d’apparition dans la séquence. Il faut donc également définir une fonction de comparaison basée sur leurs positions dans la séquence.
Les tableaux renvoyés par la fonction asort sont indexés à partir de 1 (donc ici, nous parcourons le tableau de l’indice 1 à l’indice nb inclus). Nous utiliserons la fonction delete qui permet de supprimer un élément d’un tableau sans changer son indice. Cette fonction permet également de détruire un tableau (ainsi la ligne 77 déclare implicitement la variable tmp1 comme étant un tableau). Ainsi, après avoir trié le tableau d’ORF tab1 passé en entrée par tailles décroissantes (ligne 79), il suffit pour la position i, associée à un ORF qui a été conservé jusqu’à présent (test de la ligne 81), de chercher dans les ORF suivants (boucle de la ligne 82 à 86) ceux qui sont petits et inclus dans l’ORF à la position i du tableau trié (test de la ligne 83). Ces petits ORF inclus sont tout simplement supprimés (ligne 84). Dans un second temps, il convient de compacter le tableau obtenu et de mettre à jour le compteur nb (lignes 89 à 99). Enfin, il reste à réordonner les ORF par ordre d’apparition dans la séquence (lignes 100 et 101).
Au final, il suffit d’appeler la fonction de filtrage à la fin du traitement de chaque fichier (lignes 137 et 138) avant de produire la sortie (en veillant bien à utiliser le tableau RES au lieu du tableau ORF dans les lignes 142 à 144).
En appliquant ce script à la place du précédent, on obtient un nouveau fichier décrivant 36 ORF (il ne faut pas oublier de corriger l’estimation de la taille de la séquence) :
La visualisation graphique avec annotation sketch produit une cartographie plus lisible (cf. figure 3).
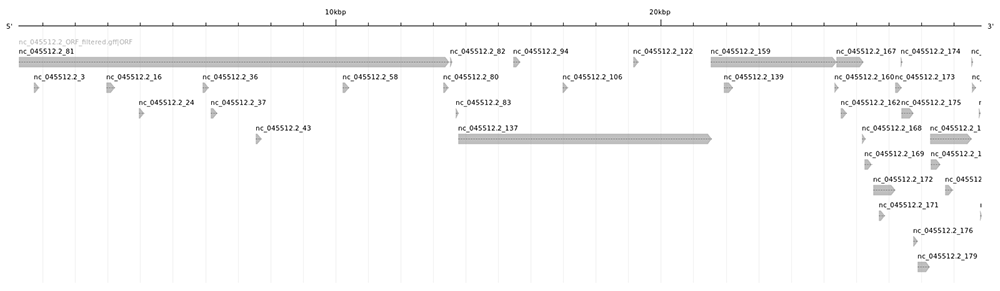
Maintenant que nous avons un nombre raisonnable de candidats (et surtout de candidats vraisemblables), nous allons pouvoir les analyser.
1.3 Récupération des séquences d'intérêt
Pour la récupération des séquences sélectionnées, il y a plusieurs solutions : ici, j'ai choisi d'utiliser seqretsplit sur le fichier contenant toutes les traductions des ORF d’intérêt, puis de ne conserver que les séquences correspondant aux identifiants voulus (un peu bourrin, mais efficace). Nous aurions tout aussi bien pu utiliser un programme du type agrep.
Nous sommes contents, nous avons isolé des régions d’intérêt, mais il reste à savoir en quoi elles sont intéressantes.
2. Étude approfondie des régions d’intérêt de la séquence de référence
Une manière assez simple de chercher si une séquence biologique est pertinente et sa fonctionnalité est de chercher s’il existe d’autres séquences proches et ce que l’on sait sur elles. Par analogie, il m’arrive parfois (trop souvent) d’avoir un doute sur l’orthographe ou le sens d’un mot, voire son existence. Dans ce cas, je me réfère systématiquement au dictionnaire de l’Académie française [7]. Cette démarche est tout à fait raisonnable et légitime dès lors que la base de données consultée est fiable, fournie et accessible.
Pour être plus précis, nous allons chercher parmi les séquences référencées dans la base de données celles qui pourraient potentiellement correspondre à ce que l’on appelle des gènes orthologues (cf. encadré).
Gènes orthologues
Vous avez certainement entendu dire de nombreuses fois que l’Homme descend du singe et que le singe descend du poisson (voire de l’arbre selon les personnes). Eh bien, rien de tout cela n’est vrai, sauf éventuellement les singes qui descendent des arbres.
Les grands singes et les hommes – au sens d’espèces – auraient un ancêtre commun qui aurait vécu il y a environ 30 millions d’années et l’ensemble des primates actuels – dont le savant homme (homo sapiens) fait partie – aurait un ancêtre commun qui aurait vécu il y a environ 60 millions d’années. Outre les nombreux conditionnels de cette phrase, ce qu’il faut retenir, c’est que la notion d’ancêtre commun est une hypothèse de travail qui permet de remonter à l’ancêtre ultime (LUCA de son petit nom pour Last Universal Common Ancestor) qui serait physiquement assez proche d’une amibe.
Acceptons cette hypothèse et engageons-nous en plein milieu d’une saga médiévale fantastique, occulte et complotiste. Le génome (comprendre ici l’ensemble des gènes) des organismes évolue au fil du temps. Certaines mutations sont délétères et les mutants ne survivent pas. D’autres mutations vont au contraire donner des capacités nouvelles à nos amies les amibes : comme avoir un nez permettant donc de porter des lunettes ou encore la possibilité de se reproduire à deux plutôt que tout seul. Mais là, je m’égare…
Dans l’idée des gènes orthologues, supposons que le génome d’une espèce E vivant à une époque lointaine possède un gène donné, alors plusieurs milliers d’années plus tard, les espèces descendantes de E peuvent avoir plus ou moins conservé ce gène. Les versions dérivées du gène ancestral sont appelées des gènes orthologues (tout ça pour ça !).
Bien évidemment, la séquence codant pour le gène peut avoir été altérée, mais la fonction du gène bien conservée, ou inversement la séquence peut avoir été très peu altérée, mais les modifications intervenues au fil du temps ont pu changer drastiquement son rôle fonctionnel. Comme dans tout problème difficile, on posera une seconde hypothèse de travail (donc fausse) pour le simplifier : les séquences nucléiques codant pour des gènes orthologues sont similaires (ce concept ne sera pas défini ici, car il faudrait au moins un chapitre entier juste pour conclure qu’il y a une infinité de définitions de la similarité et qu’aucune n’est pleinement satisfaisante ; ce faisant, je viens de vous faire gagner la lecture de plusieurs chapitres de thèses :-/ ).
Et pour celles et ceux qui souhaitent en savoir davantage, sachez qu’il existe d’autres modèles plus ou moins subtils d’évolution des espèces qui sont probablement tout aussi faux.
Donc en résumé, si une séquence est très similaire à la séquence d’un gène connu d’une espèce voisine (dans un modèle d’évolution crédible), il y a de fortes présomptions que la séquence code pour un gène orthologue à celui de l’espèce voisine, et donc il est vraisemblable que la fonction du gène soit conservée.
En bioinformatique, il existe plusieurs bases de données, l’une des plus connues est celle hébergée par le NCBI [8] qui propose pour la consulter un outil ancestral, mais toujours efficace et surtout très reconnu, j’ai nommé blast [9]. Cet outil est tout à fait installable sur un système GNU/Linux, mais ce qui le rend véritablement intéressant est essentiellement la base de données qui est interrogée, donc nous utiliserons la version en ligne. Pour y accéder, une fois sur le site du NCBI, il faut cliquer sur le bouton Blast for proteins (cf. figure 4).

Commençons par analyser le premier ORF (et accessoirement le plus grand). Il faut donc téléverser le fichier results/ORF/nc_045512.2_81.fasta.
Concernant la base de données, nous allons sélectionner la base UniProtKB/Swiss-Prot(swissprot), car cette base est constituée exclusivement de protéines annotées à la main (avec un processus de validation considéré par la communauté comme garantissant une très grande qualité/pertinence des annotations) et sans redondance (ce qui évite les biais d’interprétation liés aux comptages).
Il faut bien évidemment supprimer les éventuels résultats connus de SARS-CoV-2 puisque l'idée est de découvrir le « nouveau » virus.
La capture de la page de soumission devrait ressembler à celle présentée en figure 5.
Si d'aventure nous souhaitons ne sélectionner que certains organismes (ou les exclure), il est possible de le faire également sur la page de résultats (zone de filtre sur la droite de l'écran, cf. figure 6), puis en appuyant sur filter.
Nous obtenons 100 séquences (limite par défaut), triées par défaut par e-value (c'est le nombre de séquences similaires que nous pouvons espérer obtenir par hasard, cf. figure 7).
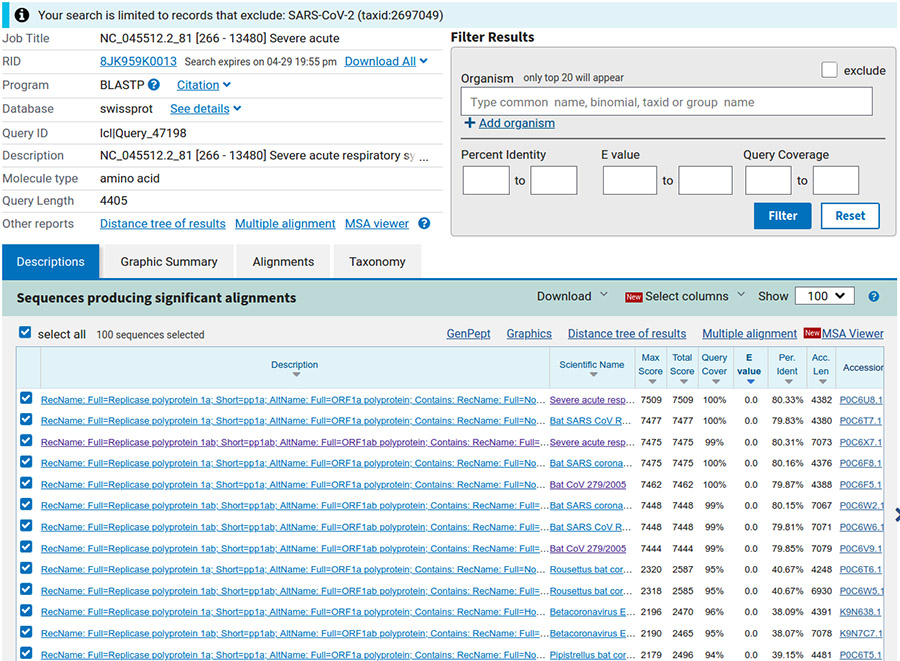
Étonnamment, les plus proches séquences trouvées proviennent de virus de la famille des coronavirus, liés à un syndrome respiratoire aigu, infectant visiblement la chauve-souris (bat en anglais). Sélectionnons les séquences ayant une couverture de notre séquence (requête) de plus de 80 % (tri décroissant au niveau de la colonne query cover). Nous obtenons 27 séquences que nous téléchargeons (menu déroulant download) au format GenBank dans le fichier results/ORF/nc_045512.2_81_related.gb.
Nous effectuons un contrôle rapide pour voir à quoi cela correspond :
Cela semble cohérent.
En cliquant sur le lien graphic summary, nous obtenons un résumé graphique (cf. figure 8) illustrant une synthèse des alignements et des annotations retrouvées dans tout ou partie des 27 séquences.
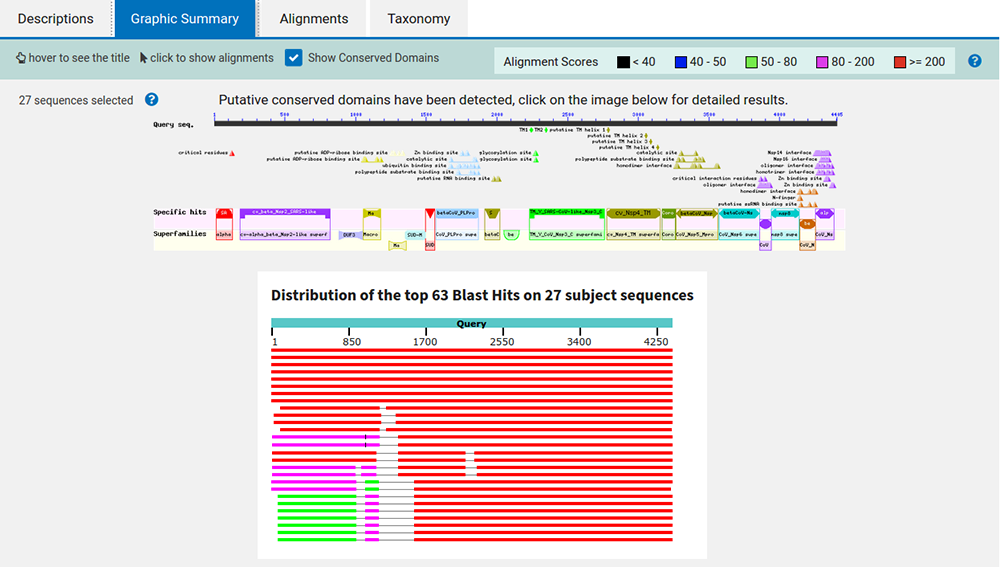
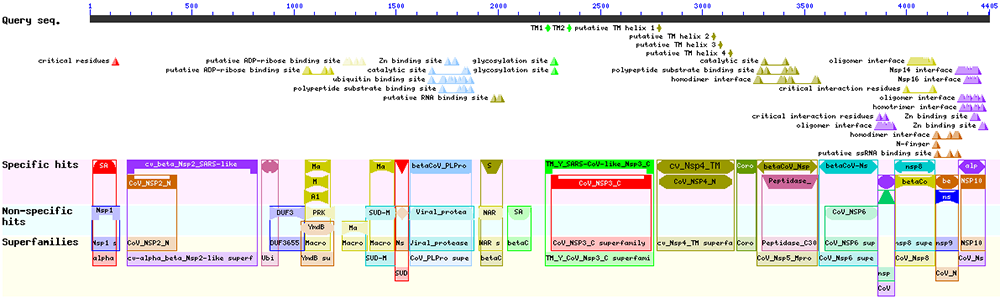
En cliquant sur l'image du haut, nous obtenons le détail des informations récupérées à partir des organismes proches (cf. figure 9).
Créons à partir du fichier que nous avons téléchargé un ensemble contenant les 27 séquences, auquel nous ajoutons la séquence qui nous a servi de requête (nous ne supprimons pas le répertoire results/tmp tout de suite, car nous en aurons besoin ultérieurement) :
Cet ensemble va nous servir à procéder nous-mêmes à l’alignement multiple avec le programme ClustalΩ. Pour cela, nous avons au moins 2,5 solutions sous Debian/Ubuntu :
- installer et utiliser le package clustalo ;
- installer et utiliser le package clustalw ;
- installer le package clustalw et utiliser le wrapper fourni avec EMBOSS.
Devinez quoi ? J’ai choisi de passer par le wrapper EMBOSS qui s’appelle emma. Donc, il faut installer clustalw :
Puis, il suffit de lancer l’alignement sur le fichier contenant les 28 séquences.
Le programme ClustalΩ construit au passage un dendrogramme (arbre de proximité/de guidage) au format newick (également appelé New Hampshire format). Il existe plusieurs outils permettant de visualiser cet arbre (figtree, treeviewx, njplot, etc.).
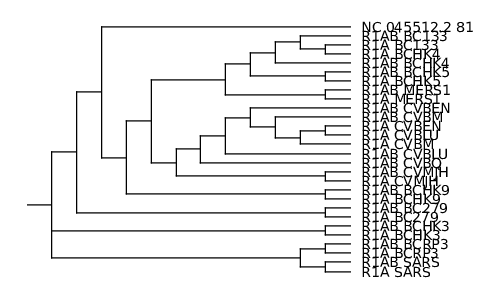
Dans un premier temps, je vous propose de tester treeviewx. C’est un outil qui nécessite de passer par une interface graphique. Il suffit de charger le fichier (pensez à changer le filtre de noms de fichiers par défaut en choisissant le format CLUSTALX guide tree (*.dnd)). Le programme permet d’exporter l’arbre affiché au format svg, ce que j’ai fait. J’ai ensuite converti l’image au format PNG grâce à l’outil convert (fourni avec le package imagemagick). Le résultat est l’image de la figure 10.
Je vous propose également d'utiliser njplot. Si son interface graphique très rétro (Tcl/Tk) est assez peu engageante, le paquet fournit également deux programmes assez sympathiques: newicktotxt et newicktops. Ces deux outils prennent le nom du fichier newick en paramètre et produisent respectivement un fichier ASCII ou Postscript représentant l'arbre. Le nom du fichier est construit à partir de la base du fichier newick avec l'extension .txt ou .ps (attention la base s'arrête au premier . rencontré et non au dernier).
L’arbre affiché par njplot n’est pas rigoureusement identique à celui proposé par treeviewx (je ne suis pas spécialiste et je ne tenterai donc pas de l'expliquer). Celui produit par njplot me semblant a priori plus cohérent, et l’outil permettant de faire de la ligne de commande, c’est celui qui je conserve au final (l’image est disponible sur le dépôt Git).
EMBOSS propose également des outils permettant de travailler sur les alignements de séquences ; il est par exemple possible de récupérer des informations sur l'alignement ou bien de produire des images permettant d'avoir une vue globale du résultat (l’image est également accessible sur le dépôt Git) :
En regardant de plus près le fichier d'alignement (results/ORF/nc_045512.2_81_related.emma), nous nous apercevons qu'aux environs de la position 5 580, les séquences dont le nom commence par R1AB_ continuent l'alignement tandis que les séquences commençant par R1A_ et la séquence requête s'arrêtent. Ceci explique l'aspect de l'image obtenue avec plotcon (results/ORF/nc_045512.2_81_related_plotcon.1.png). Lorsque nous regardons l'arbre de guidage (cf. figure 10), nous nous apercevons également que les séquences préfixées par R1AB_ sont très proches (voire appariées) à la séquence dont le nom est identique au préfixe R1A_ près.
Regardons de plus près à quoi correspondent ces séquences et récupérons pour cela les annotations disponibles pour ces séquences :
Les séquences ayant le préfixe R1AB_ correspondent toutes à une protéine appelée Replicase polyprotein 1ab, tandis que celles ayant le préfixe R1A_ correspondent toutes à une protéine appelée Replicase polyprotein 1a. La littérature ne semble pas proposer de protéine Replicase polyprotein 1b, mais [10] explique que lorsque la séquence R1A_ est suivie d'un ORF valide, le produit de cet ORF peut se combiner visiblement avec la chaîne de la protéine Replicase polyprotein 1a pour former la protéine Replicase polyprotein 1ab (je passe les détails techniques qui n'ont pas d'intérêt ici). Ceci explique que blast ait renvoyé les deux séquences, car sur la partie correspondant à la requête, le score calculé est globalement le même. Refaisons la même analyse en ne considérant que les séquences de préfixe R1A_ :
Les images produites (disponible sur le dépôt Git) confortent dans l'idée que cet ORF que nous étudions code pour la protéine Replicase polyprotein 1a (pour information, c'est une sorte de super protéine qui permet aux virus qui en disposent de se répliquer).
Sur la base du fichier results/nc_045512.2_ORF_filtered.gff, créons le fichier results/nc_045512.2.gff que nous allons étoffer (pour chaque ligne modifiée ou ajoutée, nous utiliserons perso comme information de la seconde colonne). Nous ajoutons quelques informations triviales (pour des biologistes et celles et ceux qui ont lu le petit détour vers la biologie de [1] ;-) ) :
- bien que l'entête du fichier contienne la méta-information de la position de début et de fin de la séquence, il est d'usage d'ajouter une annotation reprenant cette information en indiquant dans la dernière colonne à quoi correspond la séquence complète ;
- il est également d'usage d'indiquer les parties précédant et suivant la traduction de la séquence d’ARN. Avant le début de la première séquence codante, la région s'appelle 5'-UTR et après la dernière séquence codante, la région s'appelle 3'-UTR. L'abréviation UTR provient de l'anglais untranslated region et le 5' (resp. 3') correspond au sens de lecture (gauche vers droite, resp. droite vers gauche) de la séquence nucléique (le numéro fait référence au numéro de l'atome de carbone utilisé lors de la synthèse des acides nucléiques) ;
- nous pouvons également ajouter l'information sur la queue poly-A.
Ce premier ORF que nous venons d'analyser semble bien être une séquence codante, aussi nous changeons son type de ORF à CDS (Coding Data Sequence) et supprimons également le score (qui était égal à la longueur de l'ORF, donc qui n'est pas véritablement un score). Nous ajoutons également dans la dernière colonne les informations que nous avons retrouvées plus ou moins à l'identique dans les 14 autres séquences, à savoir que locus_tag=1a, que product=Replicase polyprotein 1a et que note=pp1a%3B ORF1a polyprotein (je vous l’accorde, reproduire sans tout comprendre est assez discutable, mais pour tout comprendre, il faudrait faire un trop gros détour vers la biologie). En général, l'annotation d'une CDS est associée à l'annotation d'un gène. Cela passe par l'ajout du nom du gène (information sémantique), mais pour les outils d'affichage, il faut expliciter le lien de parenté avec l'annotation de gène. Ici, je triche un peu (parce que la littérature que je ne citerai pas est vraiment concordante) et j'ajoute gene=ORF1a. Nous pouvons ajouter également l'annotation du gène en dupliquant l'annotation de la CDS, et en changeant l'identifiant (j'ai juste remplacé le suffixe _81 par _ppa1, et le nom du gène ORF1a par ppa1). Ceci nous permet d’ajouter le lien formel de parenté entre la CDS et le gène en ajoutant l'information Parent=nc_045512.2_pp1a dans l'annotation de la CDS, ce qui donne un fichier d’annotation (results/nc_045512.2.gff) déjà plus précis :
Il reste à faire la même chose pour les 35 ORF restants… dans le prochain et dernier article de cette initiation.
Conclusion
Nous venons de voir comment les outils et les bases de données issus de la bioinformatique permettent de transférer de la connaissance sur de nouveaux organismes. Cela est clairement lié à l’idéologie du logiciel libre (open source) de l’ouverture des données (open data) qui est présente depuis de nombreuses années (au moins 30 ans) au sein de la communauté bioinformatique. En cela, cette science se rapproche plus de l’informatique que de la biologie, qui est encore très marquée par l’idée de breveter à tout va. Pourtant, une large majorité des biologistes estiment que la bioinformatique n’est qu’un prolongement de la biologie. Je suis certain de ne pas être un biologiste et pourtant je suis bioinformaticien. Je reste persuadé que lorsqu’un sujet d’étude ne trouve pas sa place dans un microcosme établi, c’est que le sujet ne présente aucun intérêt ou bien au contraire qu’il mérite une place à part entière. De même qu’il y a un demi-siècle, l’informatique a été admise comme une science à part entière et non plus considérée comme une extension des mathématiques, de même que la biochimie n’est ni de la biologie ni de la chimie (et donc pas de la bio‑chimie), la bioinformatique n’est ni de l’informatique ni de la biologie (et donc pas de la bio‑informatique). Cela n’en fait pas pour autant une science rivale, mais au contraire une science complémentaire, dont les fruits contribuent à l’essor de ses pairs.
Pour aller plus loin
Si vous souhaitez découvrir les problématiques spécifiques à la bioinformatique, je vous invite à découvrir et explorer le site https://rosalind.info/, que j'ai découvert très récemment. J'en profite pour remercier M. Favreau, lecteur de GLMF pour son retour sur le premier article de la série et pour m'avoir indiqué cette ressource.
Références
[1] A. MANCHERON, « Voyage initiatique vers la bioinformatique : les premiers pas », GNU/Linux Magazine France n°251 : https://connect.ed-diamond.com/gnu-linux-magazine/glmf-251/voyage-initiatique-vers-la-bioinformatique-les-premiers-pas
[2] P. RICE, I. LONGDEN et A. BLEASBY, « EMBOSS: The European Molecular Biology Open Software Suite », Trends in Genetics n°16 (6), 2000, p. 276 à 277.
[3] Site officiel de la suite EMBOSS : http://emboss.sourceforge.net/
[4] Site officiel de la Sequence Ontology (SO) : http://www.sequenceontology.org/
[5] RFC de la syntaxe générique des Uniform Resource Identifier (URI) : http://www.rfc.fr/rfc/fr/rfc3986.pdf
[6] Site officiel de la suite GenomeTools : http://genometools.org/
[7] Dictionnaire en ligne de l’Académie française (9e édition) : https://academie.atilf.fr/9/
[8] Site du National Center for Biotechnology Information (centre national américain) : https://www.ncbi.nlm.nih.gov/
[9] S. F. ALTSCHUL, W. GISH, W. MILLER, E. W. MYERS et D. J. LIPMAN, « Basic Local Alignment Search Tool », Journal of molecular biology, n°215 (3), 1990, p. 403 à 410.
[10] J. ZIEBUHR, « The Coronavirus Replicase, Coronavirus Replication and Reverse Genetics », Current Topics in Microbiology and Immunology, n°287, 2005.



