

Nul n’est besoin de convaincre que les systèmes GNU/Linux sont un atout majeur dans l’évolution des connaissances de nombreuses disciplines. C’est particulièrement le cas dans l’étude et la compréhension du vivant, qui a permis l’essor ces dernières décennies de cette « trans-discipline » qu’est la bioinformatique. Voyons à travers cette série de 3 articles comment les outils existants pour notre OS favori permettent d’appréhender la découverte d’un nouvel organisme.
Non, ne partez pas sous prétexte que dans « bioinformatique », il y a « bio » ! Soyez rassurés, nous garderons une casquette très « computationnelle » et les quelques notions de biologie qui seront présentées le seront sous un jour très simplifié (et discutable pour un véritable biologiste). L’objectif ici est avant tout de montrer à quel point les outils développés pour Linux sont efficaces pour analyser des données issues du vivant et pourquoi pas, vous faire découvrir un champ disciplinaire qui n’est ni tout à fait de l’informatique, ni tout à fait de la biologie.
Prenons un exemple très concret : un virus visiblement assez contagieux pour l’homme semble se répandre sur la planète. Les communautés scientifiques s’en inquiètent et éprouvent donc le besoin d’étudier ce nouvel organisme. Dans un monde parallèle, Linus Torvalds et Richard Stallman n’ont pas existé ou bien n’ont pas œuvré pour la libération de l’informatique. Game over ! Nous sommes tous contaminés sans comprendre pourquoi. Fort heureusement, dans notre monde bien réel, nous avons de nombreux outils accessibles, exploitables, libres d’utilisation... Au moins, nous saurons pourquoi !
Après avoir récupéré quelques données, nous allons commencer par prendre en main les outils de la suite EMBOSS [1,2], puis nous analyserons la séquence virale. Bien évidemment, nous comparerons nos résultats avec l’état actuel des connaissances. Au passage, nous aurons montré (je l’espère) une fois de plus l’intérêt et l’importance de la reproductibilité de la science.
Les données récupérées, les résultats obtenus et les scripts développés dans le cadre de cette série d’articles sont disponibles sur la forge logicielle GitLab hébergée par Framasoft https://framagit.org/doccy/SARS-CoV-2-bioinfo-analysis.
1. À la découverte de la suite EMBOSS
Le nom EMBOSS est simplement l’acronyme de « The European Molecular Biology Open Software Suite ». D’une certaine manière, tout est dans le titre. C’est européen ; c’est du logiciel libre ; ça sert en biologie moléculaire (car en fait il y a plusieurs champs disciplinaires en biologie, certains pensent même que la bioinformatique est une sous-discipline de la biologie – quelle rigolade).
L’idée sous-jacente est de regrouper dans cette suite des outils simples, qui s’utilisent en ligne de commande et qui permettent de manipuler les données dans différents formats, de récupérer des données à travers Internet... le tout avec pour philosophie que « EMBOSS breaks the historical trend towards commercial software packages » (ça, c’est envoyé !).
Formats de fichiers
La notion de format utilisée dans le cadre de cette série article (et de la suite EMBOSS) renvoie ici à la structuration de fichiers texte, utilisant exclusivement le codage ASCII de base. Les extensions des fichiers sont donc utilisées à titre informatif afin de décrire la structuration du fichier ou bien pour rappeler quel programme en est à l’origine. Par exemple, un fichier d’extension .csv (pour Comma Separated Value) est un fichier texte, tel que chaque ligne se lit comme une succession de valeurs séparées par des virgules ; a contrario, un fichier .monprog est un fichier qui aura été généré par le programme monprog.
Dans sa conception, tous les outils de la suite utilisent les mêmes noms (avec la même sémantique) pour les options des lignes de commande. Certains outils tiers sont également rendus disponibles à travers des wrappers (programmes faisant office d’interfaces) permettant leur utilisation avec la syntaxe d’EMBOSS.
1.1 Installation
Commençons par installer EMBOSS. Pour les chanceux dont la distribution propose le paquet, le plus simple reste de passer par le gestionnaire ad hoc. Par exemple, sur une distribution basée sur Debian (dans mon cas, une Ubuntu 16.10), c’est :
Pour les moins chanceux, la page de téléchargement du site officiel [2] explique tout très bien. Il faut d’abord récupérer les sources (soit le lien direct vers l’archive, soit via le dépôt CVS), et si c’est une première installation, cela passe par :
Si c’est une réinstallation, alors cela passe par :
Au vu de l’ancienneté du projet et du fait qu’il est toujours maintenu et activement exploité par la communauté, je doute qu’il y ait un quelconque problème lors de l’installation.
1.2 Récupération des données
Avant toute chose, commençons par structurer nos répertoires. Dans le répertoire principal que j’ai nommé analysis, j’ai choisi de créer un répertoire pour les données récupérées, un répertoire pour les résultats obtenus et un répertoire pour les scripts que nous allons écrire.
Pour commencer, allons sur le site du NCBI [3]. Sur le bandeau (cf. figure 1), il y a un lien vers les données relatives au SARS-CoV-2 (qui nous emmène à https://www.ncbi.nlm.nih.gov/sars-cov-2/).

De là, il faut choisir d’explorer dans le hub (cf. figure 2), puis sélectionner la vue tabulée.
![]()
Au moment de la rédaction, il y a 298 215 séquences disponibles. Nous n’allons pas toutes les récupérer. Le menu de gauche propose de filtrer les résultats. Premièrement, nous n’allons conserver que les séquences complètes (filtre intitulé Nucleotide completeness). Nous allons ensuite filtrer par région géographique et ne conserver que les résultats provenant de Chine (pour avoir la séquence originelle) et de France (parce que « cocorico »). Enfin, nous allons nous replonger à la date du 16 mars 2020 et ne conserver que les séquences issues d’échantillons prélevés avant cette date (filtre intitulé Collection Date ; attention, les dates sont à fournir dans le format MM/DD/YYYY). Normalement, il reste 189 séquences candidates.
Nous sélectionnons la séquence NC_041255 (qui a une petite étiquette RefSeq indiquant que c’est la séquence de référence), la séquence MT023538 (qui est la première séquence collectée en France à avoir été accessible), et au moins une autre séquence au hasard, histoire d’en avoir trois (j’ai choisi la MW301121, qui est la dernière collectée en Chine à avoir été accessible, mais vous pouvez vraiment prendre celle que vous voulez).
Il reste à les télécharger (bouton Download, laissez les options par défaut pour les fenêtres qui s’ouvrent). Enregistrons le fichier dans le répertoire analysis/data/ sous le nom proposé par défaut (sequences.fasta).
Nous voici parés pour prendre en main la suite EMBOSS !
1.3 Exploration
En suite très complète, EMBOSS propose également des outils permettant de découvrir justement la suite. Le premier outil à prendre en main est celui qui permet de rechercher un outil par mot-clé de sa description. Si aucun mot-clé n’est indiqué, alors tout le catalogue est affiché.
Impressionnant, n’est-ce pas ? Près de 350 programmes à portée de main, dont deux autres programmes que nous allons pouvoir tester très rapidement : seealso et tfm.
Eh oui, comme leurs noms le laissent supposer, le premier indique des programmes en lien avec celui donné en argument, tandis que le second vous donne accès à « The F***ing Manual ».
Vous pouvez bien évidemment tester ou lire les documentations de tous les programmes. Parmi les options communes à tous, il y a deux options que l’on retrouve systématiquement : l’option -auto, qui valide les valeurs de chaque option fournie ou non (par défaut, les programmes vous donneront toujours la possibilité de modifier ou de confirmer les valeurs de chaque paramètre principal) et l’option -stdout qui affiche le résultat du programme sur la sortie standard. Ainsi, il est possible de vérifier que le fichier que l’on a téléchargé précédemment contient bien 3 séquences (avec seqcount), on peut récupérer quelques informations comme leurs noms et leurs longueurs (avec infoseq) ou bien encore afficher l’une des séquences (avec nthseq).
Une autre option magique et omniprésente est l’option -filter qui permet de récupérer la sortie d’un programme de la suite en entrée du programme courant. Par exemple, pour récupérer le nom et la longueur de la troisième séquence il suffit de composer :
Si vous avez saisi la philosophie de la suite, vous êtes parés à continuer, sinon entraînez-vous encore un peu sans l’option -auto.
2. Premières analyses
Maintenant que nous avons vu les rudiments des outils de la suite, commençons à explorer les séquences que nous avons récupérées.
2.1 À chacune son fichier
Ce sera assez court, car pour créer un fichier par séquence, il suffit d’une instruction :
Ainsi, il est possible de comparer deux séquences entre elles en les alignant (c’est-à-dire en essayant de faire correspondre au mieux leurs parties communes).
2.2 Alignement global
Un des algorithmes les plus connus est celui de Needleman & Wunsch [4], basé sur le principe de la programmation dynamique. Il est bien évidemment implémenté dans la suite :
Le fichier produit (results/nc_045512.2-mt320538.2.needle) permet de visualiser les points communs (et donc les différences) entre les deux séquences.
On peut notamment remarquer qu’il y a 6 différences entre les deux séquences, en plus de la variation du nombre de A à la fin de la chaque séquence (on appelle cette suite de A la queue poly-A).
Il est possible de changer le format de sortie (option -aformat3 ; le format par défaut de needle étant srspair). Les formats markx1 et markx2 sont dans le cas présent les plus lisibles (avis subjectif).
Pour ces 2 séquences, il est possible d’obtenir le même résultat d'alignement, mais beaucoup plus efficacement, avec stretcher.
Il existe un autre outil qui lui va focaliser sur les différences, et qui permet de retrouver la même information que précédemment :
Cet outil produit trois fichiers. Le premier (results/nc_045512.2-mt320538.2.diffseq) montre les différences entre les deux séquences :
Les deux autres fichiers (results/nc_045512.2.diffgff et results/mt320538.2.diffgff) focalisent sur les variations de chaque séquence par rapport à l’autre, qui est alors considérée comme une référence. Ces deux fichiers sont dans un format appelé GFF (sur lequel nous reviendrons dans le prochain article) qui n’est autre qu’un fichier tabulé, comme illustré par le second fichier généré :
2.3 Un petit détour vers la biologie
Tout d’abord, pas de panique. L’objectif ici n’est pas de vous préparer pour le bac, mais simplement de vous fournir le minimum requis pour comprendre la suite des analyses que nous allons faire. Si vous êtes férus de biologie, disons que je vous propose une autre vision de la discipline. Si vous avez un (ou une) ami(e) biologiste, c’est également l’occasion de le (ou la) faire pleurer des larmes de sang.
Imaginons une citée médiévale du nom de Kêr-kellig avec son mur d’enceinte. Au cœur de cette ville, un temple où est concentrée toute la connaissance dans des milliers de parchemins. Chaque parchemin est bien archivé dans de grandes armoires, et pour éviter toute perte irrémédiable, chaque exemplaire dispose d’une sorte de négatif permettant de le reproduire rapidement. Autour de ce temple vivent des commerçants, des artisans, des gens d’armes... Lorsqu’un artisan doit par exemple réparer un élément d’une bâtisse ou en fabriquer un, il va avoir besoin d’une partie des connaissances précieusement conservées dans le temple. Il n’est pas envisageable un seul instant que l’un des précieux manuscrits sorte des murs du temple. Aussi dans ce bâtiment, de nombreux érudits travaillent à en faire des copies. Bien évidemment, il n’est pas question d’utiliser les pigments rares et la même qualité de vélin que pour les manuscrits originaux, car une fois sortie du temple, l’artisan se permettra de rayer les parties inutiles de sa copie et lorsque celle-ci ne lui sera plus nécessaire, il l’enverra au recyclage (Kêr-kellig est une citée médiévale avant-gardiste qui recycle ses déchets). Les pigments et les parchemins pourront ainsi servir à nouveau pour produire de nouvelles copies.
Cette cité imaginaire est calquée sur le fonctionnement des cellules eucaryotes. Le temple de Kêr-kellig en est le noyau qui contient l’ensemble des chromosomes (les manuscrits). Un manuscrit est donc métaphoriquement une séquence d’ADN (Acide DésoxyriboNucléique). Chaque lettre du manuscrit s’appelle nucléotide et est composée de trois parties : au centre de cette structure, il y a un pentose (un sucre composé de 5 atomes de carbones numérotés de 1 à 5 et qui a perdu un atome d’oxygène, d’où son nom de désoxyribose) ; fixée au carbone numéro 1, il y a une des quatre bases nucléiques possibles (Adenine, Cytosine, Guanine ou Thymine, identifiées par leur initiale) ; enfin, un groupement phosphate relie le carbone numéro 5 du nucléotide au carbone numéro 3 du nucléotide suivant de la séquence (un peu comme le fait de relier les lettres avec l’écriture cursive). Cette numérotation des carbones confère un sens de lecture, non pas de la gauche vers la droite, mais du carbone numéro 5 vers le carbone numéro 3. Il est alors question des extrémités 5’ et 3’ et du sens 5’-3’.
De même que les précieux manuscrits originaux ne sortent jamais du temple et qu’en informatique, on ne travaille jamais sur un original mais sur une copie, les séquences d’ADN des chromosomes ne sortent jamais du noyau. Au besoin, ces séquences sont transcrites (copiées) en utilisant non plus des acides désoxyribonucléiques (ça coûte cher d’enluminer), mais des Acides RiboNucléiques (ARN), qui sont certes moins stables, mais coûtent moins cher à produire (du point de vue énergétique). Les sucres composant les nucléotides sont ici des riboses, et les bases nucléiques sont également au nombre de 4. On retrouve l’adénine, la cytosine et la guanine, mais la thymine est remplacée par l’uracile. Ces considérations étant essentiellement liées à la biochimie, nous considérerons que T et U, c’est pareil (larmes de sang :’( ). Comme dit le méchant M. Brochant dans « Le dîner de con » : « il est un peu râpeux c’est vrai, mais sincère, je l’ai à très bon prix » ; mais revenons à nos copies que l’on appelle ARN messagers. Ils véhiculent un message et contiennent également des éléments supplémentaires, à l’instar d’un courrier postal (ou d’une trame réseau) qui est embarqué dans une enveloppe. Ainsi, le message stricto sensu inclus dans cette séquence est appelé séquence codante ou CDS (Coding Data Sequence). En amont de la séquence codante (donc du côté 5’), il y a une zone qui par conséquent n’est pas traduite et que l’on appelle région 5’-UTR (pour UnTranslated Region). De même, en aval de la séquence codante (donc du côté 3’), il y a la région appelée... (je vous laisse deviner). Très généralement, la fin de cette zone est composée d’une succession d’adénosines (le nucléotide dont la base est une adénine) et il est alors question de queue poly-A. Tous ces éléments sont illustrés sur la figure 3.

La séquence codante s’appelle ainsi, car à l’instar d’un programme informatique, elle est contient un code qui permet à la machinerie cellulaire de produire une chaîne d’acides aminés (nous considérerons que « chaîne d’acides aminés » = « protéine » [-> re-larmes de sang]). Le code est vraiment simple à expliquer : à chaque combinaison de trois nucléotides, que l’on appelle codon, correspond un acide aminé. Il suffit donc de disposer de la table des correspondances, appelée table des codons, pour traduire le message codé avec des nucléotides en séquence d’acides aminés. Et comme le dit cette fois-ci Perceval dans « Kaamelott » (Livre III, « Poltergeist ») : « Le code c'est "le code" ? Ça va, ils se sont pas trop cassé le bonnet, pour l'trouver celui-là ! ».
Ainsi, étant donné le code d’une part et la CDS d’autre part, il y a trois phases de lecture possibles pour tenter de produire une séquence d’acides aminés. Algorithmiquement, ça donne :
Il se trouve que l’ADN dans les noyaux des cellules se comporte un peu comme un système RAID 5 à 2 disques (rappelez vous, le temple dispose de son propre mécanisme de sauvegarde). En effet, les nucléotides A et T sont complémentaires (un peu comme 00 et 11), de même que les nucléotides C et G (comme 01 et 10). On pourrait dire qu’ils s’emboîtent bien. Ainsi, l’ADN du noyau est présent en deux brins complémentaires (les deux disques du système) qui s’assemblent en formant la fameuse double hélice reproduite dans tous les films parlant de près ou de loin d’ADN ou de génétique. Ce qui est important ici, c’est que les deux brins sont assemblés par complémentarité, dans le sens contraire l’un de l’autre. Ainsi, étant donnée une séquence dont on ignore si elle provient d’un brin ou de son complémentaire inversé, pour tenter de la décoder, il faut également envisager une lecture dans l’autre sens (3’ vers 5’) des codons complémentaires. Par exemple, le codon ATG sur un brin donnerait le codon CAT sur l’autre brin (ATG à l’envers, donne GTA, et le complémentaire de GTA est CAT).
Ceci revient à dire qu’étant donnée une CDS et une table des codons, sans autre information, il existe 6 possibilités de traduction (3 phases par brin). Ceci étant, pour qu’un code soit valide, il doit nécessairement commencer par un codon spécifique (qui sera traduit) et se terminer par un autre codon spécifique (qui ne sera pas traduit). On appelle respectivement ces codons les codons start et stop. Dans le cadre de l’ADN humain (et de la très grande majorité des organismes), il n’y a qu’un seul codon start : ATG (qui code pour la méthionine) et 3 codons stop : TAA, TAG et TGA. Il y a au total 20 acides aminés, alors qu’il y a 43 (= 64) codons possibles. Tous les codons codant pour un acide aminé (ou un codon stop), il y a donc en moyenne trois codons possibles pour un acide aminé donné. La fonction de codage n’est donc pas bijective, mais seulement surjective.
Analyser une séquence à la recherche d’une traduction possible consiste donc à rechercher ce que les biologistes appellent un cadre ouvert de lecture ou ORF (Open Reading Frame) valide, c’est-à-dire une séquence débutant par un codon start et se terminant par un codon stop.
2.4 Visualisation des ORF
Algorithmiquement, il n’est pas très difficile de rechercher la présence d’ORF. EMBOSS propose différents outils permettant de les rechercher ou de les visualiser. La question peut tout de même se poser de savoir si cela a du sens de rechercher des ORF sur une séquence virale. Eh bien, la réponse est oui, car précisément le virus utilise la machinerie cellulaire de l’hôte qu’il infecte pour se répliquer ; or, sa réplication passe par la production de protéines. Par contre, pour savoir quelle table de codons utiliser pour un virus, il faut savoir quelle est celle utilisée par l’organisme hôte. Dans le cas présent, l’hôte est l’humain. Donc, c’est la table standard.
Nous pouvons afficher facilement à l’écran les ORF valides :
Il est également possible de générer un fichier au format PNG (par exemple) :
L’image produite montre clairement que les grands ORF sont sur les trois phases de lecture du brin + (cf. figure 4). C’est assez cohérent, car le SARS-CoV-2 est un virus à ARN simple brin (comprendre disque dur à pas cher et pas de configuration RAID pour la sauvegarde).
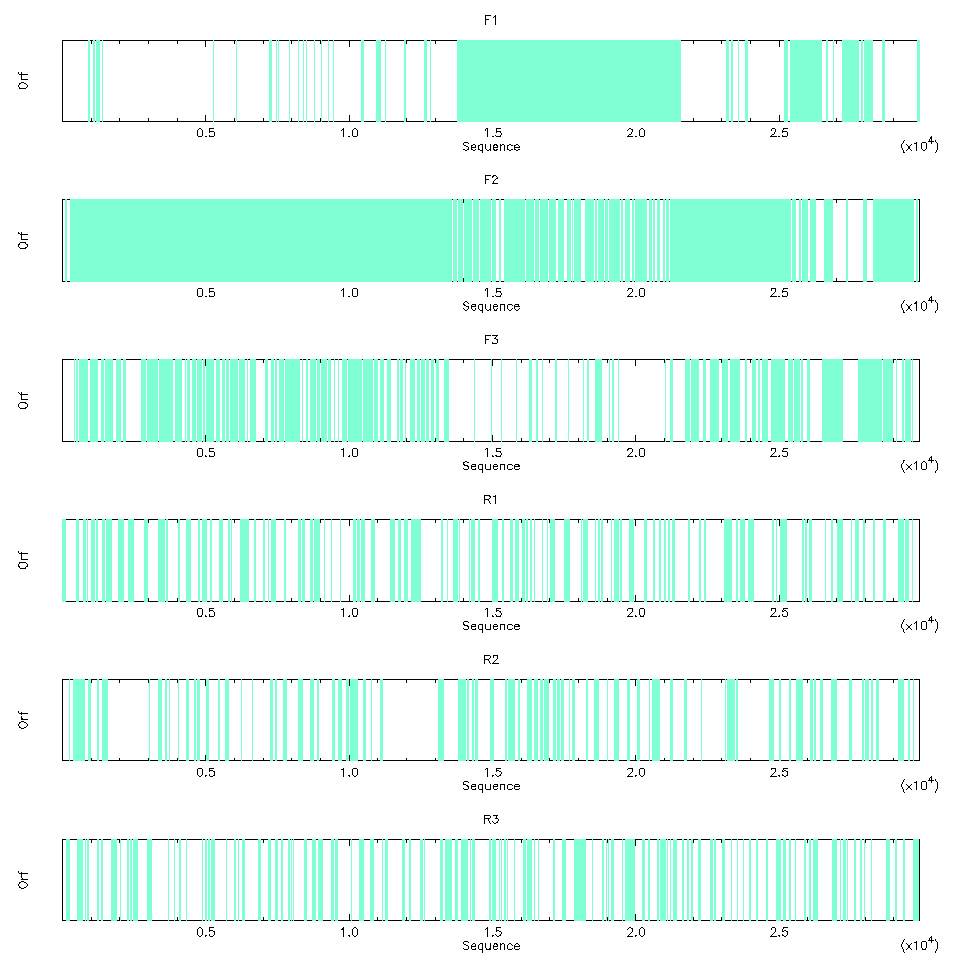
Il est aussi possible de visualiser les ORF en mode texte, ce qui permet d’avoir un peu plus de détails (nous en profitons pour limiter la visualisation aux trois phases du brin + avec l’option -frame) :
Les acides aminés qui résulteraient de la traduction sont représentés par une seule lettre et les astérisques indiquent la présence d’un codon stop.
Maintenant que nous savons qu’il y a des ORF, il ne reste pour ainsi dire qu’à les récupérer pour pouvoir les analyser.
2.5 Récupération des ORF valides
Pour récupérer les ORF valides, rien qu’une petite commande :
Le fichier produit (option -outseq results/nc_045512.2_ORF.fasta) contient les 192 traductions des ORF considérées comme valides (option -find 1) sur le brin principal (option -noreverse).
Chacune des séquences a un identifiant composé du nom de la séquence et d’un identifiant allant de 1 à 192. Nous retrouvons les positions de début et de fin de l’ORF sur la séquence d’origine et la description complète de la séquence d’origine. Comme l’hôte du virus est l’humain, nous n’avons pas spécifié de table des codons alternative, aussi toutes les séquences commencent bien par une Méthionine (codon start). Seul le dernier ORF (n°192) n’est en réalité pas valide, car il ne s’achève pas sur un codon stop, mais va jusqu’à la fin de la séquence, dont la queue poly-A, si elle était traduite, produirait une succession de lysines (K). Donc en réalité, il n’y a « que » 191 ORF valides à analyser. Ce sera l’objet des prochains articles.
Conclusion
La suite EMBOSS illustre à merveille à la fois le potentiel que représentent les systèmes GNU/Linux (en offrant un véritable outil exploitable par tous), et à la fois l’incidence de la philosophie sous-jacente aux logiciels libres dans l’avancée sur le chemin de la connaissance. À travers cette initiation, cela fait partie des messages que cet article voulait porter. Il ne s’agit pas seulement de prôner la diffusion et le partage, mais également les aspects moraux et éthiques des contributeurs de ces outils qui les mettent à disposition sans demander autre chose que d’être cités, sans demander de droit de regard sur les données analysées ou générées. Et ceci est d’autant plus important qu’en bioinformatique, ces données peuvent se révéler très sensibles (pour celles et ceux qui ne voient pas le problème, je vous invite à voir ou revoir le film « Bienvenue à Gattaca » réalisé par Andrew Niccol en 1997).
Et parce qu’il est toujours bien de finir sur un ultime rebondissement, je vous livre ici quelques noms de programmes de la suite EMBOSS qui pourraient bien vous aider au quotidien : xmlget, nohtml, noreturn, nospace et notab.
Références
[1] P. RICE, I. LONGDEN et A. BLEASBY, « EMBOSS: The European Molecular Biology Open Software Suite », Trends in Genetics n°16 (6), 2000, p 276 à 277.
[2] Site officiel de la suite EMBOSS : http://emboss.sourceforge.net/
[3] Site du National Center for Biotechnology Information (centre national américain) : https://www.ncbi.nlm.nih.gov/
[4] S. B. NEEDLEMAN et C. D. WUNSCH, « A general method applicable to the search for similarities in the amino acid sequence of two proteins », Journal of Molecular Biology n°48 (3), 1970, p 443 à 453.
Pour aller plus loin
Vous pouvez également découvrir et tester quelques-uns des outils de la suite EMBOSS en ligne, par exemple sur le serveur de l’Institut Européen de Bioinformatique (https://www.ebi.ac.uk/Tools/emboss/) ou encore sur le site de l’INRA de Toulouse (http://emboss.toulouse.inra.fr/cgi-bin/emboss). Vous pouvez également installer votre propre instance d’emboss-explorer sur vos serveurs.

{kind=link}