

Avec l’avènement de la biologie moléculaire au XXe siècle, l’homme a pris conscience qu’il pouvait utiliser le vivant pour l’étudier, puis pour le modifier. Le défi du XXIe siècle va porter sur l’utilisation intelligente de ce savoir pour accélérer l’évolution, et produire des nano machines, les protéines, capables de corriger tous les problèmes que nous avons engendrés. Mais pour cela, il faut répondre au problème le plus complexe de l’univers...
Les protéines sont de petites entités moléculaires qui jouent un rôle essentiel dans le fonctionnement des cellules. L’ADN est le support transmissible de l’information génétique (le code biologique, par analogie avec le code source en informatique), mais c’est la protéine (le binaire) qui va permettre le fonctionnement de la cellule. La difficulté de comprendre en détail ce fonctionnement vient du fait qu’il est très onéreux de déterminer la structure d’une protéine, car à ce jour, seulement 150 000 protéines ont été caractérisées (sur 150 millions identifiées). Cet effort de caractérisation des macromolécules biologiques a néanmoins permis de comprendre la plupart des grands principes d’organisation des protéines, il reste à les mettre en œuvre de manière robuste pour répondre aux grands défis du 21e siècle. C’est dans ce domaine que l’équipe DeppMind de Google a fait un grand saut en avant avec sa solution AlphaFold.
Les protéines sont des assemblages de petites briques moléculaires appelées acides aminés. Par une liaison simple et orientée, il est possible d’obtenir des agencements tridimensionnels très variés, et la nature a pu expérimenter pendant plusieurs milliards d’années pour optimiser des fonctions adaptées à la grande diversité des besoins des cellules et des organismes.
Le défi à venir pour le 21e siècle est de comprendre, puis de reproduire, comment une protéine peut-elle conduire à une fonction bien définie (si nous réussissons ce tour de force en utilisant les dernières avancées technologiques, alors il sera par exemple possible de nettoyer tout le plastique des océans ou encore, de réduire la quantité de CO2 dans l’atmosphère).
1. La difficulté algorithmique
Chaque protéine est composée d'une brique élémentaire, l'acide aminé (AA), qui peut interagir avec l'environnement (l'eau, le sang, les ions, etc.) ou avec un autre acide aminé. Il existe 20 briques élémentaires, ce qui fait que pour une séquence de deux acides aminés, il y a déjà 400 possibilités de séquences et plus généralement, 20N séquences en fonction du nombre N d'acides aminés. Pour la séquence d'une petite protéine (100 AA), cela représente par exemple déjà plus de 10130 combinaisons de séquences... Ce nombre est astronomique, car il dépasse le nombre estimé d'atomes dans l'Univers (1080). Pour prendre en compte cette complexité, Cyrus Levinthal a été le premier à estimer le temps qu'il faudrait pour qu'une séquence soit capable d'atteindre un état stable. En appliquant un raisonnement similaire à celui indiqué pour les séquences, mais en prenant cette fois-ci en compte le besoin d'agencer des acides aminés les uns à côté des autres dans l'espace, il a de nouveau estimé que le nombre de combinaisons à envisager serait compris entre 1043 et 10130… Même en imaginant que chaque combinaison puisse être testée par la nature à la vitesse d'une toutes les nanosecondes (ce qui n'est pas le cas), on arrive vite à conclure que le temps nécessaire pour le repliement d'une protéine dépasse le temps d'existence de l'Univers, ce qui est donc impossible !
Au même moment, le prix Nobel Christian Anfinsen a édicté les grands principes qui permettent d'obtenir le repliement d'une protéine dans un temps « raisonnable », compris entre la milliseconde et la seconde, comme observé expérimentalement. Ces deux grands principes sont :
- Toute l'information nécessaire au repliement d'une protéine est contenue dans sa séquence primaire en acides aminés ;
- Le repliement d'une protéine correspond à un état d'énergie minimal, un état « fondamental » qui est optimisé par la nature.
En combinant les arguments de ces deux scientifiques, il est possible de caractériser la problématique du repliement d’une protéine comme étant un problème non NP-complet, ce qui veut dire qu'il n’existe pas de solution algorithmique polynomiale que l'on peut définir. Il faut donc en conclure que ce problème doit être traité en combinant de multiples approches. Il s’agit de réduire le nombre de degrés de liberté du système (l’incertitude du modèle) à un nombre « raisonnable » qu’un ordinateur (puissant) sera capable de traiter, puisque l’approche combinatoire « brute » est impossible. Fort heureusement, nous disposons de nombreuses données biologiques « proches » sur lesquelles cette stratégie va pouvoir s’appuyer.
Les premières données biologiques fiables font appel à une combinatoire de paramètres physico-chimiques : la description des atomes des acides aminés, le calcul des interactions de ces atomes entre eux, et la prise en compte de l’interaction avec le solvant. Cette description virtuelle d’un système atomique s’appelle la mécanique moléculaire, et une version générale de ces interactions est présentée dans l’équation de la figure 1.
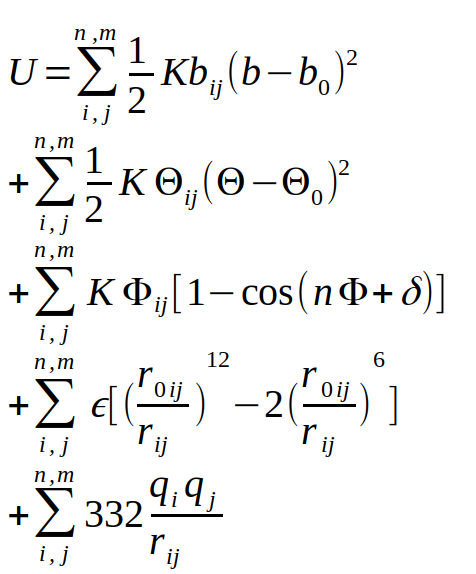
Cette description du système se base sur une description suivant les lois de Newton du monde, ce qui permet d’éviter une description quantique du système, encore impossible à calculer de nos jours pour une protéine dans son ensemble. Comme vous pouvez le constater, cette équation comprend de nombreux termes dépendants du couple d’atomes i et j considérés. Il faut donc établir des poids pour chacun d’entre eux, l’ensemble de ces paramètres s’appelle un champ de force. Comme cette description est multiparamétique (très large), il existe différentes implémentations dans des logiciels comme AMBER, CHARMM, NAMD, GROMACS ou LAMMPS, pour ne citer que les plus populaires d’entre eux. Cette description des lois de modélisation des systèmes biologiques a aussi été mise en place à la fin des années 1960, ce qui a amené les pionniers du domaine, Michael Levitt, Arieh Warshel et Martin Karplus à recevoir le prix Nobel de Chimie en 2013 [1].
L’utilisation de la mécanique moléculaire et d’un champ de force approprié s’avère très utile lorsque l’on arrive à une solution proche de l’optimum replié d’une structure, car il permet un calcul rapide et précis de l’énergie potentielle de cette structure, utile pour comparer plusieurs solutions « proches » entre elles. À l’heure actuelle, il n’est cependant pas possible de l’utiliser pour partir d’une structure non repliée (linéaire) des acides aminés pour arriver à une forme compacte de la protéine. En plus des approches théoriques indiquées précédemment, on se heurte aussi à des contraintes pratiques, comme des problèmes de convergence des calculs, ou des erreurs liées à la précision forcément limitée (même en 64 bits) des représentations numériques.
Les approches actuelles sont donc beaucoup plus pragmatiques : puisqu’il n’y a pas de solution mathématique, physique ou informatique « exacte » au problème, il est possible d’accumuler plusieurs données intermédiaires qu’il va falloir combiner, pour diminuer le nombre de degrés de liberté du système et ainsi, améliorer la fiabilité des prédictions.
L’approche la plus performante est la comparaison entre une structure à modéliser et une structure « proche », qui est par exemple disponible chez un autre être vivant avec lequel on peut établir un lien par analyse phylogénétique. Si un lien de « parenté » existe, on parle alors de modélisation par homologie, par exemple si on découvre un animal qui n’est pas encore caractérisé, mais qui possède 4 pattes, des poils, des oreilles pointues et qu’il est agile, on pourra en déduire que c’est un félin, et donc le modéliser sans le connaître exactement. Si la proximité est moins évidente, par exemple si on n’est capable de déterminer que la parenté « éloignée » entre les protéines, alors on parle de modélisation par comparaison. Dans ce cas-là, on pourrait imaginer découvrir un nouveau mammifère qui aurait comme caractéristiques de marcher sur ses 4 pattes, d’avoir une queue et une respiration aérienne avec des poumons. Dans ce cas, on perçoit rapidement qu’il y aura des zones qui risquent d’être moins bien modélisées que d’autres.
Quand on ne trouve pas de structure assez « proches », on va utiliser une approche par « enfilage » (threading en anglais), où l’on va utiliser des morceaux de protéines pour modéliser une autre protéine, un peu à la manière de Frankenstein… Cette approche a déjà été décrite ainsi par le passé par Kosinski et ses collègues [2].
La dernière catégorie de modélisation des protéines est celle qui s’appelle de novo / ab initio. Dans cette catégorie, de loin celle qui regroupe le plus grand nombre de protéines, il n’y a pas assez d’informations tridimensionnelles pour modéliser de grands segments de protéines, il faut donc presque à chaque fois la modéliser à partir de zéro. Jusqu’à maintenant, les meilleurs succès ont été obtenus pour des protéines dont la taille est inférieure à 100 AA, et seules quelques modélisations ont donné des résultats probants.
Pour chacune des catégories, il est donc nécessaire de prendre une information partielle de structure (la forme d’une protéine en 3D), de la combiner avec d’autres informations hétérogènes de séquence (la suite des acides aminés est toujours connue), avec encore d’autres approches issues de différentes méthodes de prédiction, ayant chacune d’entre elles un taux de succès relativement moyen (au mieux 80 % de bonnes prédictions à l’heure actuelle).
Nous allons maintenant voir comment les derniers développements dans le domaine de l’intelligence artificielle ont permis de sérieusement revisiter le domaine.
2. L’innovation apportée par AlphaFold
Comme le problème à optimiser est complexe, il n’y a pas de solution exacte mathématiquement qui peut être appliquée directement. L’approche employée par tous les scientifiques travaillant dans ce sujet est donc de combiner des informations expérimentales de nature diverse (séquence, structure, fonction) avec des données de prédiction ayant une performance variable. Parmi ces méthodes, on peut citer deux grandes approches : Rosetta [3] et I-TASSER [4]. Dans ces approches, la séquence linéaire d’acides aminés à modéliser est « décorée » par des annotations fonctionnelles et structurales, si elles existent. Une partie d’une séquence va par exemple être reconnue comme étant une enzyme (un ciseau moléculaire), une autre partie de la séquence va plutôt se voir attribuer le rôle de liaison à un composant cellulaire, par exemple à une membrane, et ainsi de suite. À ce jour, il est estimé que l’on ne connaît réellement pour le moment que 4 % des structures des protéines dont on est capables de détecter la présence. Ces deux approches sont donc limitées par :
- la capacité à correctement détecter une analogie entre la séquence à modéliser et un fragment de protéine dont on connaît la structure ;
- la diversité des structures disponibles pour arriver à une forme tridimensionnelle exacte.
Par analogie avec l'informatique, c’est comme si l’on ne disposait pour construire un ordinateur que des composants électroniques élémentaires (condensateurs, résistances, silice), sans plan de montage… Dans ces conditions, on comprend bien qu’il est important d’avoir un exemple sur lequel se baser. Pour notre ordinateur, on aurait déjà 3 grands principes d’organisation des composants : pour un ordinateur fixe, pour un ordinateur portable ou encore, pour un serveur. Si l'on ne dispose pas de carte mère, le canevas d'assemblage de tous les composants, le problème va vite se révéler impossible à résoudre...
2.1 Une première avancée majeure : l'analyse des contacts distants entre acides aminés
Pour une protéine, comme il n’existe pas assez de composants pour servir d’exemples et/ou de canevas précis, il est possible de traiter le problème par la statistique : pour chaque acide aminé, il faut calculer la proportion que celui-ci soit présent dans une conformation en hélice, en feuillet, ou indéterminée. La réponse à cette prédiction est une probabilité conditionnelle dont on ne connaît pas la portée a priori. Pour un acide aminé n par exemple, cela veut dire que son état dépend de l’état de l’acide aminé n-1 et n+1, mais aussi de la position d’un acide aminé présent beaucoup plus loin dans la séquence, par exemple à la position n-25 ou n+32. Ce domaine de recherche s’appelle la prédiction de contacts [5]. Les approches classiques dans ce domaine ont consisté à isoler dans les séquences des protéines les acides aminés qui mutent « par paires », ce qui indique qu’ils sont proches dans l’espace, même s’ils sont distants dans la séquence (distance n à n+1 >> 5). Grâce aux approches de génomique à haut débit, il est en effet possible d’obtenir la séquence primaire des acides aminés à grande échelle, même pour des organismes dont on ne connaissait pas l’existence auparavant (voir à ce sujet le projet Tara Océans, par exemple). Ces séquences sont assemblées par alignement multiple et il est possible de déduire la probabilité de mutation de chaque acide aminé à une position donnée, puis de comparer ce taux de mutation avec un autre acide aminé. Si ces deux taux de mutation sont identiques, alors ces acides aminés sont appariés : quand le premier mute, le second est aussi observé muté, en compensation de la conservation de l'interaction entre ces deux acides aminés. Compte tenu de la complexité des statistiques liées à ces grands nombres, des corrections des biais d’analyse sont appliquées pour éviter les faux positifs et les faux négatifs [6]. Cette approche avait permis récemment de franchir un pas dans la qualité de la prédiction de la structure des protéines, l’équipe de Rosetta ayant en effet réussi en 2017 à prédire correctement la structure de plus de 70 % des protéines qu’il fallait découvrir.
2.2 Quelle a été l'avancée apportée par DeepMind ?
Les créateurs de AlphaFold, la version spécifique de l'équipe de DeepMind appliquée au problème de la prédiction de structure des protéines, ont capitalisé sur le travail de leurs prédécesseurs. Parmi les auteurs du travail, on peut par exemple citer David Jones, expert dans le domaine de la prédiction de structures de protéines, et expert dans le domaine de la prédiction de motifs à l'aide de réseaux de neurones (voir à ce sujet l'article sur la prédiction de structures secondaires de protéines dans le HS n°94). Comme indiqué ci-devant, nous ne connaissons actuellement la structure tridimensionnelle que pour 4 % des protéines. Ce nombre représente tout de même plus de 29 000 structures, quand on enlève la redondance entre les structures résolues expérimentalement (plus de 150 000 disponibles dans la Protein Data Bank ou PDB [7]).
En utilisant les technologies développées par David Jones (HHblits) et sa connaissance de la structure des protéines, les ingénieurs et chercheurs de Google ont pu mettre en place un protocole « simple » de prédiction :
- filtrer et nettoyer les données de structure et de séquence des protéines pour avoir un jeu de données de référence pour l'apprentissage,
- définir une stratégie « simple » de prédiction des interactions, en se focalisant sur la prédiction des contacts entre acides aminés proches et lointains (jusqu'à 63 acides aminés de distance) et sur la prédiction des angles des carbones bêta des acides aminés (ceux qui indiquent l'orientation des chaînes latérales des acides aminés).
Pour ceux qui ne sont pas familiers avec les acides aminés et/ou la structure des protéines, vous pouvez consulter les vidéos explicatives de Siraj Raval [8] et plus complexes par Andrew Senior (de Google) [9] qui présentent les concepts et les détails de l'implémentation d'AlphaFold. La stratégie « simple » de Google est développée à partir de 29 minutes et 30 secondes dans la vidéo de Andrew Senior.
La grande avancée de AlphaFold a résidé en l'utilisation de convolution expansée (convolution dilated [10]), appliquée de manière itérative. En dilatant ainsi l'espace bidimensionnel de mesure des contacts connus, issus des interactions observées dans la structure des protéines présents dans la PDB, ils ont ainsi obtenu un prédicteur très efficace des contacts potentiels entre deux acides aminés. Même si la performance de ces approches et le code exemple de leur travail n'est pas disponible, on peut percevoir à travers les communications publiques disponibles que la puissance de calcul qui a été utilisée a représenté plusieurs années en équivalent mononœud (ils ne parlent même pas de temps CPU unique)… Chaque paramètre du réseau de neurones et sa structure ont été estimés de nombreuses fois, pour obtenir des prédictions de grande qualité, supérieures à ce qui a été fait par les concurrents du domaine. En « dilatant » ainsi l'espace des conformations, ils ont pu indiquer au réseau de neurones quels étaient les paramètres les plus importants pour la structure des protéines, sans entrer dans les détails physico-chimiques qui sont traditionnellement pris en compte par les approches plus traditionnelles des équipes de recherche. Grâce à cet apprentissage, ils ont ainsi pu retrouver des propriétés pour lesquelles David Jones avait réalisé il y a vingt ans des prédicteurs dédiés !
Ces deux matrices de distance entre acides aminés, et d'angles entre les carbones bêta des acides aminés ont servi à définir un jeu « grossier » de contraintes sur les positions des acides aminés, et la minimisation de ces contraintes a été réalisée par une approche de minimisation dite de la plus grande pente, avec l'algorithme L-BGFS [11], tel qu'implémenté dans la suite Scikit-learn développée à l'INRIA [12] (Cocorico !). Cette étape initiale a été complétée par l'utilisation du travail de Baker et de ses collègues, disponible dans la suite Rosetta, afin d'obtenir un modèle tridimensionnel final qui a été soumis pour évaluation.
3. L'évaluation des résultats
La prédiction de la structure des protéines est un domaine de recherche très actif depuis des décennies. Dans un premier temps, il a servi à prédire le modèle d'une protéine en se basant sur la structure d'une protéine proche, par exemple parce qu'il était plus simple expérimentalement d'extraire le matériel biologique d'un animal que d'un humain. La rhodopsine, la protéine qui nous sert à transformer le signal lumineux d'un photon en signal électrique pour notre cerveau, a ainsi été cristallographiée à partir d'extraits de bœuf ou de poulpe. Comme les méthodes de prédiction commençaient à faire appel à de plus en plus d'approches complexes, il a été décidé de juger à l'aveugle si les prédictions des équipes de recherche correspondaient à la réalité. La compétition CASP (Critical Assessment of Structure Prediction, http://predictioncenter.org/) a donc commencé en 1994 et la dernière édition, la 13e, a eu lieu en 2018. Le principe général est que des structures de protéines qui ont été caractérisées expérimentalement par des experts du domaine ne sont pas rendues publiques dès qu'elles sont disponibles, mais conservées « un certain temps » pour que les modélisateurs du monde entier produisent un modèle qui sera évalué. La comparaison entre la structure connue et le modèle fait appel à plusieurs critères, révisés à chaque édition, qui vont tenir compte de la position des atomes de la structure connue et mesurer la distance qui sépare chacune de ceux-ci avec le même atome de la structure modélisée. Cette comparaison interatomes est réalisée pour tous les atomes, et la mesure globale qui en résulte (RMSD) sert à déterminer si un modèle est proche ou non de la structure connue. Cette Root Mean Square Distance, ou somme des moindres écarts carrés des distances en français, doit être égale ou inférieure à 2 angstrœms (10-10 m) pour un très bon modèle, à moins de 5 angstrœms pour un modèle moyen et à une valeur encore supérieure, si le modèle est mauvais. Les protéines étant de grandes structures, une pondération par classes est effectuée pour éviter que si une partie d'un modèle est mal prédit, mais que le reste est correct, ce modèle ne soit considéré comme intégralement mauvais. Cette mesure de RMSD pondéré s'appelle le GDT_TS. Dans l'idéal, un GDT_TS de 100 % indique une correspondance parfaite, un très bon modèle sera supérieur à 75 %, un modèle intermédiaire compris entre 50 % et 75 % et les mauvais modèles auront un GDT_TS inférieur à 50 %.
Quand on compare le résultat brut de AlphaFold en prenant cette simple mesure pour l'évaluation (voir figure 2), on peut constater que l'équipe de Google a réussi un tour de force pour cette première participation : la plupart des modèles produits ont été classés comme de bons modèles et ils ont relégué le meilleur scientifique à la deuxième place, alors qu'il se classait systématiquement premier depuis plusieurs éditions de CASP. Cette méthode d'évaluation par le GDT_TS est souvent complétée par une évaluation experte humaine qui prend en compte d'autres critères pour déclarer le gagnant final et par le passé, des discussions sont apparues sur cette évaluation « humaine ». Dans cette deuxième évaluation, il n'y a cette année pas de débat, AlphaFold se classe encore à la première place, loin devant ses poursuivants !
Conclusion
Est-ce la fin de ce domaine de recherche ? Avec ses moyens incommensurables, Google a-t-il apporté un point final aux recherches dans le domaine ? Non. L'approche de Google s'est basée sur de nombreuses approches établies depuis des décennies que le blog d'AlphaFold reprend rapidement. De l'aveu même du chef de l'équipe AlphaFold, Andrew Senior, cette approche a bien fonctionné, car une attention toute particulière a été apportée à la prédiction des contacts, à la mise en place du réseau profond de neurones, et à son optimisation. Des moyens « conséquents » ont été mis en œuvre, mais sans détail précis : le nombre d'ingénieurs impliqués dans le projet n'a pas été communiqué, la puissance de calcul utilisée n'a pas été révélée, etc. En pratique, Google a prouvé que des solutions technologiques performantes étaient disponibles, et qu'en les assemblant sérieusement, il était possible de produire des modèles intéressants. Sans les logiciels de David Jones et de David Baker, il n'y aurait pas d'AlphaFold, il faut donc combiner les meilleures approches pour être bon. Cependant, bon dans ce domaine ne suffit pas, les meilleurs modèles produits par AlphaFold ont un GDT_TS de moins de 90 %. Quand il s'agit de prédictions, ce taux est très acceptable, par contre quand il s'agit de comprendre le vivant, à grande échelle, il faudra encore améliorer la performance de ces prédictions de structures, pour combler le fossé entre les 4 % de structures connues, et les 96 % à modéliser pour comprendre le vivant. Avec ces 4 %, nous avons déjà réussi à produire des médicaments pour la plupart des maladies humaines, à modifier le vivant pour corriger des défauts de fonction, nous commençons même à l'améliorer. Imaginez ce qui serait possible si on arrivait à prédire avec fiabilité ne serait ce que 20 % de plus de structures de protéines !
Références
[1] Annonce des récipiendaires du prix Nobel 2013 de Chimie : https://www.nobelprize.org/prizes/chemistry/2013/press-release/
[2] J. KOSINSKI et collaborateurs, « A “FRankenstein's monster” approach to comparative modeling: Merging the finest fragments of Fold‐Recognition models and iterative model refinement aided by 3D structure evaluation », Proteins n°53 (56), octobre 2003, p. 369 à 379.
[3] S. OVCHINNIKOV et collaborateurs, « Large-scale determination of previously unsolved protein structures using evolutionary information. » Elife. 2015 4:e09248. doi:10.7554/eLife.09248.
[4] J. YANG et collaborateurs, « The I-TASSER Suite : Protein structure and function prediction. » Nature Methods, 12: 7-8 (2015).
[5] Matrice de contacts dans les protéines : https://en.wikipedia.org/wiki/Protein_contact_map
[6] Voir à ce sujet l’explication sur le site de bioinformatics.org : http://www.bioinformatics.org/aces/
[7] Le dépôt de toutes les structures connues de macromolécules : http://www.rcsb.org
[8] Vidéo explicative de AlphaFold par Siraj Raval : https://www.youtube.com/watch?v=cw6_OP5An8s
[9] Vidéo explicative de AlphaFold par Andrew Senior : https://www.youtube.com/watch?v=uQ1uVbrIv-Q
[10] Explication de la convolution dilated : https://towardsdatascience.com/understanding-2d-dilated-convolution-operation-with-examples-in-numpy-and-tensorflow-with-d376b3972b25
[11] La méthode de minimisation dite de la plus grande pente « L-BGFS » : https://fr.wikipedia.org/wiki/Méthode_de_Broyden-Fletcher-Goldfarb-Shanno
[12] L'implémentation en Python de nombreuses méthodes de Machine Learning : https://scikit-learn.org



